
LES GRANDS SYNDROMES EN ORTHOPEDIE DENTO FACIALE
LES GRANDS SYNDROMES EN ORTHOPEDIE DENTO FACIALE
1.INTRODUCTION
- Le développement des structures craniofaciales met enjeu des mécanismes dont nous commençons à appréhender la complexité.
- L’Altération du massif cranio facial dans ses différents composants tissulaires en particulier osseux aboutissent à des malformations craniofaciales qui se déclinent en : Fentes, Dysostose et Synostose
- La connaissance des causes de ces malformations permet de mettre en relief, dans la mesure du possible, le gène responsable en premier mais aussi et surtout de comprendre de quelle manière ce gène altéré peut conduire à une déviation de la croissance et du développement physiologique.
2. DEFINITION
2-1-Définition d’un syndrome
Grec : Sindromé: concours ou réunion
Réunion d’un groupe de symptômes qui se reproduisent en même temps dans un certain nombre maladies
2-2-Définition de la génétique
du grec genno = donner naissance
La génétique est la science qui étudie les fonctions chimiques inhérentes à une espèce particulière de molécules appelée gène. Une de ses branches, la génétique formelle ou mendélienne, s’intéresse à la transmission des caractères héréditaires entre des géniteurs et leur descendance
3. LES GRANDS SYNDROMES EN ODF
3.1. LES AFFECTIONS CONGENITALES
3.1.1. Les fentes labio-paltines
Les FLP sont les malformations crânio-faciales les plus fréquentes chez l’Homme. Elles peuvent faire partie du tableau clinique de nombreux syndrome. On considère qu’elles sont d’origine polyfactorielle. Elles sont plus fréquentes sous forme isolée ou non syndromique (70 à 95 %).
Ce sont des anomalies de développement de la région maxillaire supérieure apparaissant au cours de l’embryogenèse, elles correspondent à une solution de continuité, soit au niveau de la lèvre supérieur, soit au niveau du voile du palais, soit au niveau du maxillaire, soit au niveau de toutes ces structures à la fois (dans les cas les plus graves, la fente s’étend de la narine au pharynx décrivant ainsi les variétés élémentaires suivantes:
- La fente labiale : elle intéresse la lèvre supérieure parfois jusqu’au seuil narinaire. L’aile nasale est étalée. L’alvéole est respectée.
- La fente labio-alvéolaire : elle concerne la lèvre supérieure avec une fissure de la région alvéolaire voire de tout le palais primaire. La fente se situe habituellement dans la zone du germe de l’incisive latérale. La communication bucco-nasale est rare et se fait au niveau du palais primaire. L’éruption dentaire peut être altérée dans les suites.
- La fente vélaire : elle peut aller de la luette jusqu’au bord postérieur du palais osseux. La forme minimale est la bifidité de la luette. La fente vélaire complète s’accompagne d’une incompétence musculaire du voile, d’un jetage nasal lors de la déglutition et plus tard d’une rhinolalie. Cette fente peut être sous-muqueuse visible uniquement lors d’un examen attentif.
- La fente vélo-palatine : elle intéresse le voile et le palais osseux jusqu’au foramen incisif(palais secondaire) et expose le vomer. Cette fente constitue une large communication bucco-nasale.
- Des mutations de gènes tels que TWIST et FGFR2 sont à l’origine d’anomalies cranio -faciles syndromiques sévères.
- Plusieurs gènes défectueux ont été étudiés et mis en relation avec les fentes oro-faciales (TGFa, TGFb, MSX1, MSX2, RARa…) avec des associations alléliques et des pénétrances variables.
- Chez l’homme, plusieurs tératogènes ont été mis en relation avec l’apparition de fentes faciales : tabac, alcool, rétinoïdes, aminoptérine, diphénylhydantoïnes, triméthadione, thalidomide
- Ce caractère multifactoriel rend par ailleurs le conseil génétique aux parents difficile et empirique. Un modèle d’estimation du risque familial de récurrence de la malformation a été proposé par Tolarova il ne reste qu’indicatif et doit être modulé par des variables telles que le sexe, la race, la sévérité de l’atteinte et les facteurs environnementaux.

LES GRANDS SYNDROMES EN ORTHOPEDIE DENTO FACIALE
4.1.2. Syndrome de VAN DER WOUDE.
C’est caractérisé par de petites dépressions (ou fistules) de la lèvre inférieure, de profondeur variable, associées ou non à une fente labiale, labio-palatine ou palatine. C’est le plus fréquent des syndromes avec fente orofaciale : sa prévalence dans la population générale est d’environ 1 cas pour 60 000. Une hypodontie peut être associée au VWS. Le tableau clinique peut être très variable, même à l’intérieur d’une famille, avec toutes les combinaisons possibles, mais les dépressions labiales sont présentes dans 88 % des cas. Ce syndrome se transmet sur le mode autosomique dominant, avec une forte pénétrance (80 à 97 %). Des mutations du gène IRF6 (locus 1q32-q41) ont récemment été mises en cause dans le VWS. Plus de 70 mutations ont été rapportées, affectant le fonctionnement du facteur de transcription IRF6 (interferon regulator factor 6). Les diagnostics différentiels sont les syndromes facio-génito-poplitéal (poplitealpterygium syndrome) et orofaciodigital, dans lesquels on retrouve parfois les puits labiaux paramédians, pourtant très suggestifs du syndrome Van der Woude. Le traitement des malformations du VWS concerne surtout la réparation de la fente orofaciale. Les fistules labiales sont généralement asymptomatiques mais leur excision chirurgicale peut être recommandée pour des raisons esthétiques, ou pour réduire la salivation excessive.

LES GRANDS SYNDROMES EN ORTHOPEDIE DENTO FACIALE
Signes cliniques
4.1.3. Syndrome de Crouzon
La maladie de Crouzon associe une craniosténose et une hypoplasie du massif facial. La craniosténose est variable, mais le plus souvent plusieurs sutures sont concernées. La dysmorphie faciale est caractéristique avec un hypertélorisme, un exorbitisme (lié au double recul du maxillaire supérieur et du front) et une inversion de l’articulé dentaire. Un élément important est que la synostose crânienne est évolutive : habituellement peu ou pas visible à la naissance, elle apparaît vers l’âge de 2 ans et s’aggrave progressivement. Il existe cependant des formes précoces, congénitales, dans lesquelles l’hypoplasie maxillaire supérieure est très importante et responsable de difficultés respiratoires et d’un exorbitisme majeur pouvant menacer les globes oculaires par défaut d’occlusion palpébrale. Ces formes représentent peut-être une entité à part. L’hydrocéphalie est fréquemment observée dans la maladie de Crouzon, et peut poser de difficiles problèmes thérapeutiques.
C’est un syndrome à transmission autosomique dominante, pour lequel on trouve une mutation du gène FGFR2 chez 60% des malades testés. Une forme particulière est liée en plus à une mutation spécifique (Ala 391 Glu) dans le domaine transmembranaire d’une autre protéine de la même famille, FGFR3. Le risque d’hypertension intracrânienne (avec sa conséquence majeure, lacécité) est particulièrement important, menaçant deux patients sur trois d’où la nécessité de la chirurgie.
- Très fréquent
- Turricéphalie/oxycéphalie/acrocéphalie
-
- Brachycéphalie/occiput plat
- Craniosténose
- Hypertélorisme
- Exophtalmie
- Fréquent
- Rétrognathisme/micrognathisme
- Nez en bec d’oiseau
- Lèvres proéminentes
- Palais ogival/étroit
- Dents mal implantées
- Occasionnel
- Front bombé/bosses frontales
- Mandibule absence partielle/hypoplasie
- Strabisme
- Lèvres minces/retractées
- Macroglossie
- Anodontie/oligodontie


LES GRANDS SYNDROMES EN ORTHOPEDIE DENTO FACIALE
Examen téléradiograhique:
4.1.4. Le syndrome d’APERT
C’est une malformation majeure, associant une facio-craniosténose et des syndactylies osseuses et membranaires des quatre extrémités. Son incidence est de 1 naissance sur 50 000. La craniosynostose, visible dès la naissance, est toujours bicoronale. Le système longitudinal (sutures métopique et sagittale) est anormalement large, même durant les premiers mois de vie. Le maxillaire supérieur est très hypoplasique, avec inversion de l’articulé dentaire, et la face est large, avec un nez en bec, un hypertélorisme constant et un exorbitisme parfois important. La syndactylie des doigts et orteils peut être totale (aspect en moufle des extrémités) ou partielle, affectant les deuxième, troisième et quatrième doigts. L’atteinte mentale est extrêmement fréquente et souvent lourde, souvent associée à des malformations cérébrales. La grande majorité des patients (plus de 98%) présente une des deux mutations adjacentes (Ser252Trp et Pro253Arg) du gène FGFR2 (pour fibroblast growth factor receptor type). L’insertion d’éléments Alu dans ou près de l’exon 9 de ce même gène est en cause dans les autres cas. Le risque d’hypertension intracrânienne est élevé, approchant 50% des cas. Une intervention précoce sur la craniosténose (avant l’âge de 6 mois) peut améliorer le pronostic mental.
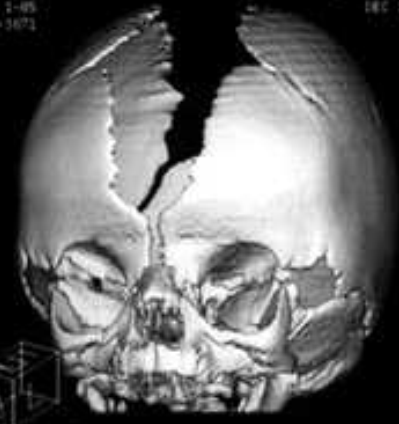


LES GRANDS SYNDROMES EN ORTHOPEDIE DENTO FACIALE
- Fréquent
- Fontanelle:large/retard de fermeture
- Hypertélorisme
- Fente palpebraleantimongoloïde
- Strabisme
- Palais ogival/étroit
- Convulsions épilepsie
- Occasionnel
- Nez mince
- Choanes atrésie
- Fente palatine
4.1.5. Syndrome de PFEIFER
Syndrome associant une craniosténose et une syndactylie partielle des mains et des pieds.
Il atteint 1 individu sur 100 000 transmissions autosomiques dominantes,
Il peut être provoqué par des mutations des gènes FGFR-1 ou FGFR-2 (fibroblast growth factor receptor).

Signes cliniques :
Occasionnel
- Face plate
- Face asymétrique (pas de paralysie faciale)
- Prognathisme
- Bouche entre6ouverte en permanence
- Philtrum court
- Palais ogival/étroit
4.1.6. Syndrome de BINDER
C’est une dyststose maxillo-nasale (syndrome naso-maxilo- vertébral):
Ce syndrome semble être de transmission anormale sur le chromosome X.
Signes téléradiographiques :


LES GRANDS SYNDROMES EN ORTHOPEDIE DENTO FACIALE
4.1.7. Le syndrome de FRANCESCHETTI et ZWAHZEN (de Treacher Collins)
Le syndrome de Treacher Collins (ou syndrome de Franceschetti-Klein) est une affection génétique qui se transmet sur le mode autosomique dominant avec une pénétrance de 90% et une expressivité variable. Son incidence est estimée à 1/50 000 naissances. Ce syndrome associe : une hypoplasie des pavillons des oreilles (77%), une atrésie des conduits auditifs externes (36%), une anomalie de la chaîne des osselets, une surdité de transmission (40%) ; une hypoplasie des os malaires et zygomatiques (80%) avec obliquité anti-mongoloïde des fentes palpébrales ; un colobome des paupières inférieures (69%) avec absence de cils du 1/3 externe de la paupière inférieure ; une hypoplasie mandibulaire (78%) ; une fente palatine (28%). Les malformations faciales sont bilatérales et asymétriques. L’intelligence est généralement normale. Des difficultés respiratoires peuvent se manifester précocement du fait de l’étroitesse des voies respiratoires supérieures. La surdité doit être dépistée le plus tôt possible.
Le taux de nouvelles mutations était estimé à 60% mais il serait en fait moindre d’après les études moléculaires récentes. Le gène est localisé sur le chromosome 5q32-q33.1.
Signes cliniques:
- Très fréquent
- Fente palpebrale antimongoloïde
- Rebord orbitaire plat
- Rétrognathisme/micrognathisme
- Menton pointu
- Fréquent
- Racine du nez proéminente
- Palais ogival/étroit

- Occasionnel
- Microphtalmie
- Choanes atrésie

4.1.8. Syndrome du premier arc ou syndrome Oto-mandibulaire

LES GRANDS SYNDROMES EN ORTHOPEDIE DENTO FACIALE
4.1.9. Le syndrome de Goldenhar
Le syndrome de Goldenhar ou dysplasie oculo-auriculo-vertébrale (OAV) est caractérisé par une hypoplasie faciale asymétrique associée à des anomalies oculaires (dermoïde ou lipodermoïdeépibulbaire, microphtalmie, colobome de la paupière supérieure…), des appendices ou sinus pré-auriculaires et des malformations vertébrales (fusion des cervicales, puzzle vertébral…). L’atteinte faciale est généralement unilatérale, mais peut être bilatérale avec une expression plus sévère d’un côté. D’autres malformations sont rapportées dans 50% des cas : anomalies cardiaques, cérébrales, rénales, gastro-intestinales. Un retard mental n’est présent que dans 10% des cas. Il existe un risque d’apnées obstructives dans le sommeil. La prévalence de la dysplasie OAV est estimée entre 1/5 600 et 1/20 000 naissances. La plupart des cas sont sporadiques et le risque de récurrence empirique est faible (2-3%). Dans les cas familiaux, le mode de transmission est compatible avec une hérédité autosomique dominante. La dysplasie OAV apparaît comme un défaut de champ de développement embryonnaire complexe.
- Signes cliniques :
- Très fréquent
- Face étroite
- Rétrognathisme/micrognathisme
- Mandibule absence partielle/hypoplasie
- Fente palatine
- Macrostomie/grande bouche
- Surdité de transmission/conduction
- Vertèbres anomalie de taille/forme
- Fréquent
- Paralysie faciale
- Pommettes plates/malaire hypoplasie
- Fente labiale latérale
- Très fréquent

4.1.10. L’achondroplasie
L’achondroplasie est la chondrodysplasie la plus fréquente et touche un enfant sur 15 000. Elle est caractérisée par une petite taille avec des membres courts, une hyperlordose, des mains courtes, une macrocéphalie et un front haut avec une ensellure nasale marquée. Ce désordre est responsable d’un déficit de taille assez sévère (taille finale 130cm +/- 10cm) et de déformations squelettiques modérées, hyperlordose, et parfois des complications neurologiques liées à l’étroitesse du canal rachidien. Le développement intellectuel est normal. Le diagnostic repose sur l’examen radiologique. Cette affection se transmet selon un caractère dominant autosomique. Cependant environ 90% des personnes atteintes naissent de parents sains : il s’agit d’une nouvelle mutation, c’est-à-dire d’accident génétique. Le gène responsable est FGFR3 qui code pour un récepteur de facteur de croissance fibroblastique exprimé dans le cartilage de croissance. Il s’agit d’une mutation unique identifiable par étude moléculaire. Le diagnostic anténatal est possible. A ce jour, les seules possibilités thérapeutiques sont d’ordre orthopédique.
Signes cliniques :
- Très fréquent
- Macrocéphalie
- Front bombé/bosses frontales
- Nez plat
- Racine du nez deprimée
- Micromélie (structure normale)
- Petite taille / nanisme
- Transmission autosomique dominante
- Fréquent
- Face étage moyen de hauteur reduite
- Narines petites / triangulaires
- Mains courtes/brachydactylie

4.1.11. La trisomie 21(syndrome de DOWN)
C’est une anomalie chromosomique définie par la présence d’un 3ème exemplaire, en totalité ou en partie, du chromosome 21. La trisomie 21 n’est pas une anomalie rare, mais son incidence à la naissance a diminué significativement dans plusieurs pays, après la mise en place du dépistage prénatal. Une déficience intellectuelle variable, souvent légère, une hypotonie musculaire et une laxité articulaire quasi-constantes sont les conséquences habituelles, souvent accompagnées de signes morphologiques et d’un risque de complications, justifiant un suivi adapté. Les particularités morphologiques (fentes palpébrales en haut et en dehors, épicanthus, nuque plate, visage rond, nez petit, pli palmaire unique bilatéral) peuvent être discrètes et ne sont pas pathognomoniques. Les principales malformations et complications possibles incluent : malformations cardiaques et digestives (atrésie duodénale), cataracte congénitale, maladie de Hirschsprung, petite taille, syndrome de West, épilepsies, leucémies, apnées du sommeil, déficits sensoriels, pathologies auto-immunes et endocriniennes (hypothyroïdie, intolérance au gluten, diabète, alopécie), vieillissement plus précoce et maladie d’Alzheimer. Dans 95% des cas, il s’agit d’une trisomie 21 libre (par non-disjonction méiotique) et homogène ; elle est en mosaïque dans 2-3% des cas. Enfin, dans 2-3% des cas, elle est non libre, c’est à dire que le chromosome ou la partie du chromosome 21 surnuméraire est intégré à un autre chromosome. Le caryotype permet de poser le diagnostic. Pour les parents d’un enfant porteur de trisomie 21 libre, le risque de récidive est peu modifié (1% jusqu’à 40 ans, lié à l’âge maternel ensuite). En cas de trisomie 21 non libre, le risque n’augmente que si l’un des deux parents est porteur d’une anomalie équilibrée. Pour une personne porteuse de trisomie 21, le risque de transmission est de 1/3. L’espérance de vie médiane est maintenant supérieure à 50 ans.
Signes cliniques :

- Très fréquent
- Brachycéphalie/occiput plat
- Face plate
- Fente palpebrale mongoloïde
- Racine du nez deprimée
- Hyperlaxité ligamentaire
- Mains courtes/brachydactylie
- Pied court / brachydactylie orteils
- Retard mental / psycho-moteur
- Hypotonie
- Trisomie ou monosomie totale/partielle
- Fréquent
- Fontanelle:large/retard de fermeture
- Nez court/nez petit
- Nez plat
- Microstomie/petite bouche
- Lèvres épaisses
- Coins de la bouche tombants
- Bouche entreouverte en permanence
- Macroglossie
- Palais ogival/étroit
- Anomalie de la dentition
- Microdontie totale ou partielle

4.1.12Syndrome de Moebius
- Le syndrome de Moebius correspond à une paralysie congénitale des muscles des yeux et du visage.
- Le premier symptôme est une difficulté à téter. Les nouveau-nés peuvent aussi baver de manière excessive et présenter un strabisme. Ensuite, c’est l’absence d’expression du visage (et de sourire notamment), et l’absence de clignements et de mouvements latéraux des yeux qui dominent le tableau clinique.
- Le syndrome de Moebius est dû à une anomalie de développement du 7ème nerf crânien (facial) dans tous les cas, et dans 75% des cas du 6ème (abducens). D’autres nerfs crâniens peuvent être affectés plus occasionnellement (notamment les 3ème, 4ème, 5ème, 9ème, 10ème et 12ème).
- La plupart des cas sont sporadiques sans histoire familiale particulière.
- Transmission autosomique dominante

Signes cliniques :
Très fréquent
- Paralysie faciale
- Noyaux des nerfs crâniens agénésie.
Occasionnel
- Rétrognathisme/micrognathisme
- Fente sousmuqueuse/luette bifide
- Lèvres proéminentes
- Microglossie/aglossie
- Palais ogival/étroit
- Microdontie totale ou partielle
- Email anomalie

4.2. AFFECTIONS ACQUISES
4.2.1. Acromégalie
L’acromégalie est le résultat d’une augmentation de la sécrétion d’hormone de croissance (somathormone GH ) par l’hypophyse. Cette augmentation de la sécrétion est en règle générale secondaire à une tumeur bénigne de l’hypophyse (adénome de l’hypophyse).
C’est une maladie rare, touchant environ 40 personnes sur un million, essentiellement des femmes de 30 à 40 ans.

Signes cliniques :
- Croissance exagérée des mains.
- Prognathie mandibulaire évolutive.
- Développement anormale de la langue.
- Macro rhinie.
- Selle turcique très ouverte

LES GRANDS SYNDROMES EN ORTHOPEDIE DENTO FACIALE
4.2.2 .Ankylose Temporo Mandibulaire
Due le plus souvent à des lésions anatomiques condyliennes avec soudure plus ou moins complète des surfaces articulaires osseuses et lésion du cartilage condylien associé à des troubles fonctionnels sévères avec limitation considérable parfois absence totale du jeu mandibulaire lorsqu’elle est symétrique elle est responsable de la classe II par brachymandibulie.

4-3-SYNDROMES CLINIQUES SECONDAIRES AUX ANOMALIES DU COMPORTEMENT NEURO-MUSCULAIRE
4-3-.1. Le syndrome Pierre-Robin
- Le syndrome Pierre-Robin ou mieux, la séquence Pierre Robin, est défini par une triade morphologique orofaciale faite d’un Rétrognathisme, d’une Glossoptose et d’une fente vélo-palatine postérieure médiane. la fente palatine postérieure est un défaut de fermeture du palais secondaire, lié à la persistance de la langue en position verticale et à un défaut de développement mandibulaire. Cette anomalie de développement mandibulaire est d’origine variée, rarement primitivement osseuse, le plus souvent secondaire à une hypo-mobilité oro-faciale anténatale, généralement par défaut de fonctionnement du tronc cérébral primitif (rhombencéphale). Ceci explique la fréquence et la gravité des signes fonctionnels néonataux, à savoir troubles de succion-déglutition-ventilation, avec tétées difficiles, fausses routes, troubles de la motricité de l’œsophage, obstruction ventilatoire glossopharyngo-laryngée, malaises vagaux.
- La séquence de Pierre Robin est isolée dans environ 50% des cas (aucune autre malformation n’y est associée). Dans ces formes isolées, on trouve environ 10% de cas familiaux.
- Aucun gène n’a été isolé à ce jour.
- Une consultation de génétique spécialisée est conseillée même dans les cas apparemment sporadiques.
Signes cliniques :
- Très fréquent
- Rétrognathisme/micrognathisme
- Fente palatine
- Glossoptose
- Fréquent
- Pharynx anomalie/déglutition anomalie
- Détresse respiratoire
- Fente sous muqueuse/luette bifide
- Lèvres proéminentes
- Microglossie/aglossie
- Palais ogival/étroit
- Microdontie totale ou partielle
- Age osseux retard
- Retard pubertaire/hypogonadisme
- Retard mental / psychomoteur
- Conduction nerveuse anormale
- Petite taille / nanisme.

4-3-.2. Syndrome de Rix:
C’est une anomalie de comportement de la zone oro-labiale déterminée par l’action modelante anormale du sphincter oro-buccal au cours de la succion infantile de « refuge »l’enfant tête anormalement, la succion domine la situation
- chez l’enfant affectif et émotif une mimique caractéristique se renouvelant à chaque période d’inquiétude,
- aspire sa joue et sa lèvre d’une manière gourmande.
- les muscles des lèvres et du menton sont fortement contractés, appuyés par les joues au niveau des commissures, agissant fortement sur processus alvéolaire.
Examen clinique :
- Déglutition atypique
- Bascule pathologique du massif lingual
- « retrognathie fonctionnelle » :
- la lumière du pharynx modifié par la malposition du massif lingual.
- La mandibule peu développée.
- Rétroalvéolie<accompagnant l’open bite ou la proalvéolie supérieure.
- Profil normal.
Examen téléradiographique :
- Massif lingual dans sa position de repos laisse le pharynx largement ouvert.
- Cet élément essentiel + position normale de la mandibule confirme le syndrome
5-3-.3. Syndrome d’Eschler (Cauhépé Fieux)
- C’est une anomalie de position linguale déterminée par une mimique de refuge à dominance latérale.
- Succion déséquilibre linguo-mandibulo-hyoidien:
Interposition latérale de la lèvre et de la langue et la déglutition devient pathologique.
Cliniquement :
- Déviation de l’étage inférieur de la face associée à des troubles d’occlusion.
Jeu mandibulaire :
- Balancement dévié du côté de la langue.
- Mandibule de forme normale.
- Articulation latérale croisée D ou G
- Déviation du chemin de fermeture

6. Conclusion
Devant la complexité et la variabilité clinique et symptomatologique de ces malformations congénitales et acquises, il parait primordial pour l’orthodontiste de connaître l’étiologie de leur survenue et le mode de transmission des affections congénitales, afin de pallier les anomalies qui leur sont associé et surtout pour prévenir leur survenue pour les générations qui suivent.
Aussi l’expression génétique ne pouvant être modifiée, la présence d’un facteur héréditaire influence lourdement le plan de traitement et le pronostic propre des patients présentant l’une ou l’autre des malformations.
LES GRANDS SYNDROMES EN ORTHOPEDIE DENTO FACIALE
Voici une sélection de livres:
- “Orthodontie de l’enfant et de l’adulte” par Marie-José Boileau
- Orthodontie interceptive Broché – Grand livre, 24 novembre 2023
- ORTHOPEDIE DENTO FACIALE ODONTOLOGIE PEDIATRIQUE
- Orthopédie dento-faciale en dentures temporaire et mixte: Interception précoce des malocclusions Broché – Illustré, 25 mars 2021
- Nouvelles conceptions de l’ancrage en orthodontie
- Guide d’odontologie pédiatrique: La clinique par la preuve
- Orthodontie linguale (Techniques dentaires)
- Biomécanique orthodontique
- Syndrome posturo-ventilatoire et dysmorphies de classe II, Bases fondamentales: ORTHOPÉDIE ET ORTHODONTIE À L’USAGE DU CHIRURGIEN-DENTISTE
LES GRANDS SYNDROMES EN ORTHOPEDIE DENTO FACIALE
Articles similaires
L’EXPLORATION EN ODONTOSTOMATOLOGIE Radiologique Biologique
L’EXPLORATION EN ODONTOSTOMATOLOGIE Radiologique Biologique Introduction La conduite diagnostique buccodentaire impose l’association d’un bon examen clinique (l’inspection et la palpation)...
Lire l'article
Maintenance parodontale
Maintenance parodontale Maintenance Parodontale Introduction La maintenance parodontale est un pilier essentiel pour garantir le succès à long terme d’un...
Lire l'article
Implant Esthétique : Le Guide Complet pour un Sourire Naturel et Durable
Implant Esthétique : Le Guide Complet pour un Sourire Naturel et Durable Vous avez perdu une ou plusieurs dents et...
Lire l'article
Leave a Reply