
Les grands syndromes en ODF / ODF
Les grands syndromes en ODF / ODF
Introduction
Les syndromes malformatifs cranio-faciaux, heureusement peu fréquents, requièrent une prise en charge pluridisciplinaire ainsi qu’un suivi de longues années, ponctué souvent d’interventions chirurgicales. Les anomalies de l’extrémité faciale surviennent au cours du développement, elles sont très nombreuses du fait de la complexité de l’embryogenèse. Quelques-unes seront décrites.
Rappel embryologique
Les cellules de la crête neurale ou CCN ont un rôle prépondérant pendant l’embryogenèse. Au cours de la fermeture du tube neural, des cellules ectoblastiques situées au niveau des bords de la gouttière neurale perdent leur statut épithélial stationnaire pour devenir mobiles et mésenchymateuses. Elles siègent en dehors du tube neural et en dedans de l’épiblaste.
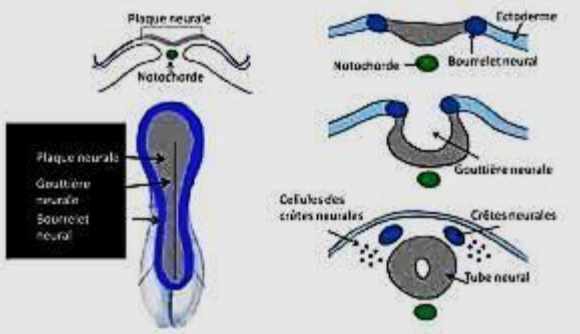
Les cellules migrent et se différencient sous l’influence de gènes. Les gènes impliqués dans le développement des CCNs sont :
- BMP (à l’exception de BMP3), Wnt, Notch, les FGFs et l’acide rétinoïque.
- D’autres gènes vont assurer le contrôle du cycle cellulaire, l’adhésion intercellulaire et les modifications du cytosquelette (la majorité des gènes ont plusieurs fonctions).
Grâce aux travaux du groupe de recherche de LE DOUARIN, il a été démontré que la crête neurale est à l’origine de nombreux dérivés céphaliques. Ainsi, tous les accidents subis par les crêtes neurales depuis leur formation jusqu’à leur différenciation, peuvent entraîner des malformations nommées de ce fait « neurocristopathies ».
Classification des anomalies faciales
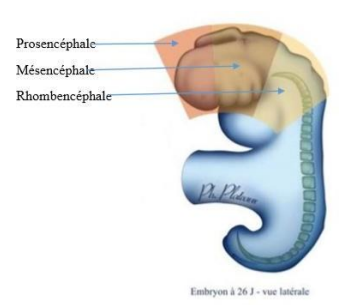
Une classification neurocristopathique est proposée et on décrit :
- Les neurocristopathies prosencéphaliques, intéressant le bourgeon naso-frontal.
- Les neurocristopathies mésencéphaliques, intéressant les bourgeons maxillaires et mandibulaires.
- Les neurocristopathies rhombencéphaliques, intéressant la région cervico-thoracique.
Les grands syndromes en ODF
Le syndrome de Crouzon
Définition / Description
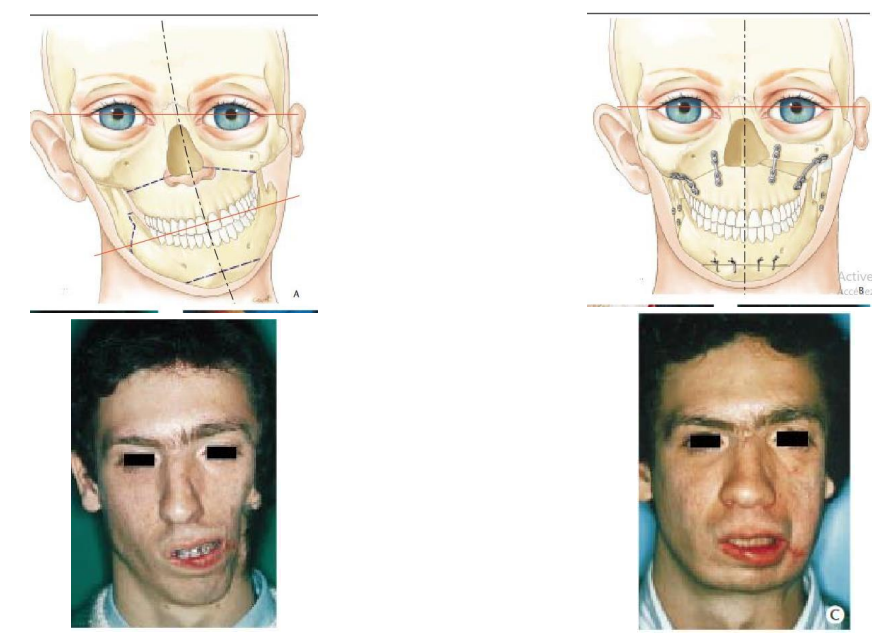
C’est une malformation cranio-faciale, d’origine héréditaire. Les signes cliniques de ce syndrome sont : une augmentation des diamètres verticaux et transversaux du crâne (acro-brachy-céphalie) et présence des impressions digitiformes sur toute la voûte crânienne. Cette craniosténose étant évolutive, le risque le plus important est l’apparition d’une hypertension intracrânienne avec cécité.
Au niveau de la face : une exophtalmie, une hypoplasie du maxillaire supérieur avec dysharmonie dento-maxillaire supérieure, endo-infra-rétrognathie maxillaire et occlusion de Classe III. Le psychisme est primitivement normal et il n’y a pas d’arriération mentale. Il n’existe pas d’atteinte des mains et des pieds. Les formes atténuées ou frustes sont possibles (expressivité variable).

Étiopathogénie
Étiologie
Elle est héréditaire, à transmission autosomique dominante, avec expressivité variable. Cette maladie serait due à des mutations dans les gènes codant pour les récepteurs de croissance (FGFR) : mutation du gène FGFR2 localisé sur le locus 26 du chromosome 10. Certaines études suggèrent que l’âge paternel augmenterait le risque de cette pathologie.
Pathogénie
C’est une neurocristopathie prosencéphalique du bourgeon naso-frontal. Elle est due à une fermeture prématurée de la suture coronale, et synostose entre le vomer et les maxillaires.
Fréquence
1 sur 200 000 naissances.
Autres noms de la maladie
- Dysostose craniofaciale type Crouzon.
- Syndrome pseudo Crouzon.
- Dysostose craniofaciale type I.
Diagnostic
Il n’existe pas de diagnostic anté-natal (échographie). Le diagnostic précoce : parfois inapparent à la naissance. L’affection se manifeste surtout, dans les premiers mois de la vie par : des altérations de la croissance crânienne (mesure des périmètres crâniens), confirmées par un examen tomodensitométrique ; des signes neurologiques et d’hypertension intra-crânienne.
Traitement
Dans l’ordre chronologique, il faut prévoir :
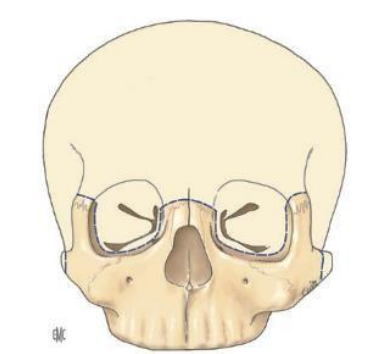
- Des interventions neuro-chirurgicales précoces : craniotomie orbito-coronale. Les buts sont la décompression et la correction de l’exophtalmie.
- Un traitement chirurgical radical de la malformation : un remodelage fronto-orbitaire par avancée et bascule du complexe orbito-naso-frontal et d’un volet frontal.
- Un traitement orthodontique : il assure la prévention des manifestations de la DDM supérieure par extractions et germectomies programmées des dents, mise en place de mainteneurs d’espaces ou des forces extra-orales (masque de Delaire). Il se poursuit par des appareils multi-attaches pour aligner les arcades et fermer les espaces d’extraction résiduels, mais en ne souhaitant pas aboutir à tout prix à une occlusion normale.
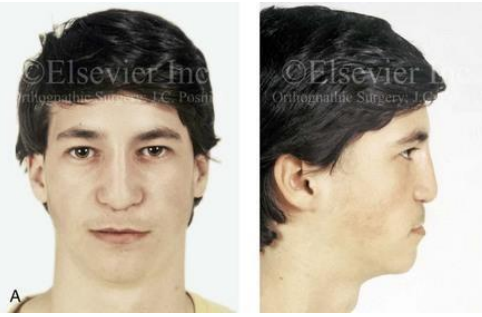
- Un traitement chirurgical orthognathique tardif « de rattrapage de la croissance » : c’est l’ostéotomie d’avancée type LEFORT I, pratiquement toujours nécessaire à la fin de la croissance, du fait de l’absence de développement des maxillaires par rapport à la croissance normale de la mandibule.
Le syndrome d’Apert
Définition / Description
C’est une craniosténose caractérisée par une soudure précoce des sutures crâniennes (sagittale et coronale). Elle intéresse le crâne, la face et les membres : « acro-céphalo-syndactylie ». Elle provoque une augmentation des diamètres verticaux et transversaux du crâne, avec présence de bosses frontales bilatérales ; une exophtalmie avec hypertélorisme, accompagnée de ptosis et de paralysies oculomotrices.
Les symptômes faciaux sont une hypoplasie maxillaire entraînant une dysharmonie dento-maxillaire supérieure avec endo-infra-rétrognathie, une prognathie mandibulaire donnant une occlusion de Classe III. La bouche est habituellement ouverte, présence d’une fissure palatine.
Au niveau des membres : syndactylies des 3e, 4e et 5e doigts et syndactylie des 3e, 4e et 5e orteils, intéressant le squelette, la peau et les ongles. Le psychisme est primitivement normal ; il n’y a pas d’arriération mentale. Il n’existe pas de formes atténuées ou frustes de ce syndrome. L’expressivité est constante. Tous les sujets atteints de syndrome d’Apert se ressemblent.
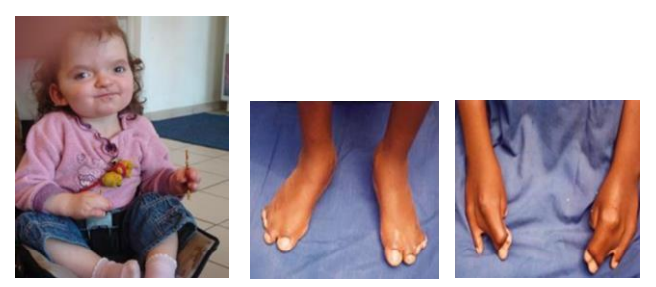
Fréquence
Il est plus rare que le syndrome de Crouzon.
Étiopathogénie
Étiologie
Il s’agit d’une maladie héréditaire, autosomale dominante avec expressivité constante, ou le résultat d’une mutation des gènes FGFR.
Pathogénie
Comme la maladie de Crouzon, c’est une neurocristopathie prosencéphalique du bourgeon naso-frontal.
Diagnostic
Il peut être anté-natal, précoce ou au cours des premières semaines de la vie, fondé sur les syndactylies ; l’hypertension intra-crânienne ; l’exophtalmie et les paralysies oculo-motrices.
Traitement
Il est assez semblable à celui de la maladie de Crouzon, il comprend en plus des interventions chirurgicales orthopédiques sur les syndactylies.
Dysplasie ou dysostose cleido-crânienne (DDC) ou Syndrome de Pierre Marie et Sainton
Définition / Description
C’est une malformation squelettique congénitale intéressant non seulement l’extrémité céphalique (face et crâne) mais aussi le reste du corps (ceintures scapulaire et pelvienne, thorax, rachis) avec un retard staturo-pondéral et des anomalies dentaires particulièrement marquées. Il s’agit essentiellement d’une polyodontie souvent considérable, intéressant particulièrement la denture permanente. L’encombrement intra-osseux provoqué par ces germes multiples entraîne des rétentions des dents permanentes et des retards de perte des dents temporaires.
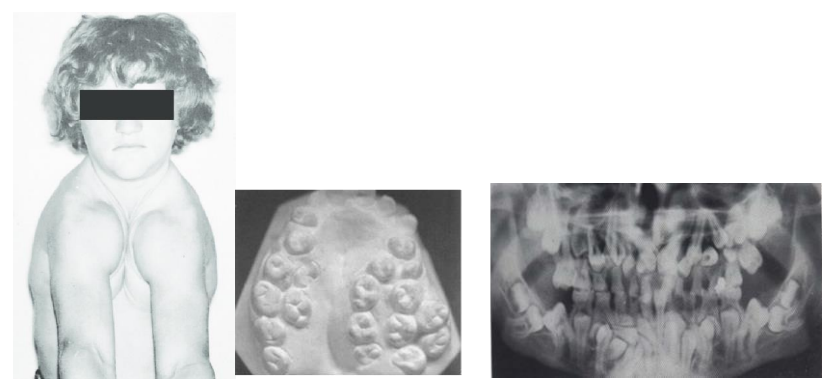
Fréquence
On ne dispose pas de données numériques concernant l’incidence de l’affection, elle est peu fréquente mais non rarissime.
Étiopathogénie
Étiologie
Il s’agit d’une maladie héréditaire selon le mode autosomique dominant, avec forte pénétrance et expressivité variable. On ne connaît pas de modifications remarquables du caryotype.
Pathogénie
Elle est mal connue parce que peu étudiée. Il s’agirait cependant très certainement d’une neurocristopathie d’origine non seulement céphalique mais aussi troncale.
Diagnostic
En l’absence de notion de diagnostic anténatal, il est parfois périnatal : retard d’ossification crânienne et larges fontanelles. Le diagnostic est en général précoce, pendant la première enfance, fondé sur les anomalies de la ceinture scapulaire et les membres. Mais parfois il est plus tardif, surtout dans les formes incomplètes, avec peu de manifestations squelettiques, s’appuyant sur des signes dentaires : persistance anormale des dents temporaires, inclusions multiples.
Traitement
Pour cette affection, on se limitera, au traitement de la polyodontie et de l’hypoplasie du massif facial.
- Le traitement de la polyodontie dépend de l’âge de la première consultation. Il consistera tout d’abord à retirer les dents temporaires persistantes, les dents surnuméraires déjà évoluées. En un ou plusieurs temps la germectomie des dents surnuméraires facilement accessibles, ce qui va pouvoir laisser l’évolution spontanée des germes « normaux ». Si l’éruption des germes « normaux » ne se produit pas leur mise en place se fera par les moyens chirurgico-orthodontiques.
- Le traitement de l’hypoplasie du massif facial se fait à l’aide des ostéotomies d’avancée de la face (l’ostéotomie du type LE FORT III).
Syndrome de Binder ou Syndrome naso-maxillo-vertébral ou Dysostose maxillo-nasale
Définition / Description
C’est une malformation héréditaire de la partie médiane de la face dérivant du bourgeon naso-frontal. Elle comporte : la réduction ou l’absence de l’angle naso-frontal, l’absence de relief de la glabelle ; l’hypoplasie et la verticalisation des os propres du nez : nez court ; racine aplatie ; absence d’arête et atrophie de la muqueuse nasale. Delaire décrit la réduction ou l’absence de l’épine nasale antérieure. On retrouve également une hypoplasie ou une absence de relief de la columelle, et un aspect en « coup de hache » de la région sous-nasale, du fait de la normalité de la partie inférieure de la lèvre supérieure.
Les signes cliniques oro-faciaux regroupent une hypoplasie médio-faciale : hypomaxillie ; classe III dentaire et squelettique.
Fréquence
L’incidence de la malformation, qui semble assez exceptionnelle, est inconnue.
Étiopathogénie
Étiologie
La transmission n’est pas connue. Certaines études ont cependant montré des formes familiales suggérant une transmission autosomique récessive ou dominante à faible pénétrance.
Pathogénie
C’est une neurocristopathie prosencéphalique : trouble de la migration, de l’induction ou de la multiplication des cellules issues de la crête neurale du prosencéphale, qui vont contribuer à la formation du bourgeon naso-frontal.
Diagnostic
Dans la majorité des cas, le diagnostic est posé au cours de la seconde enfance devant les troubles morphologiques et l’anomalie occlusale.
Traitement
Chez l’enfant ou le jeune adolescent : traitement orthopédique par tractions extra-orales postéro-antérieures sur masque orthopédique de Delaire, associé à une rééducation des fonctions linguales. Chez le grand adolescent et chez l’adulte, les sutures faciales ne peuvent plus répondre aux tractions orthopédiques. On doit alors envisager des ostéotomies d’avancée de l’étage supérieur de la face (LE FORT II ou LE FORT III).
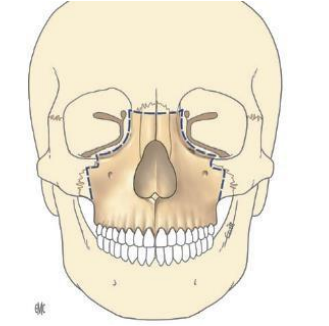
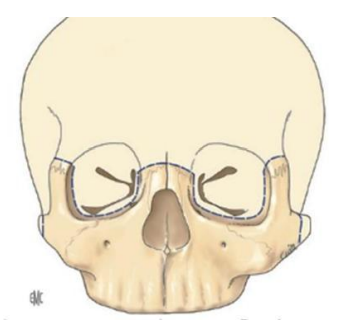
Syndrome oto-mandibulaire
Définition / Description
C’est une agénésie ou une hypoplasie bilatérale ou, le plus souvent, unilatérale, des dérivés du premier arc branchial. Quand l’atteinte est bilatérale, elle réalise le syndrome de Franceschetti-Treacher Collins. L’hypoplasie atteint les tissus suivants :
- Osseux : hypoplasie de la branche montante de la mandibule, du condyle, de l’articulation temporo-mandibulaire, du tympanal, du marteau, de l’enclume, de la caisse du tympan ; une hypoplasie du malaire et du zygoma.
- Cartilagineux : agénésie ou hypoplasie du conduit auditif externe et du pavillon de l’oreille.
- Musculaire : hypoplasie des muscles : temporal et masséter.
- Dermique : hypoplasie du derme latéro-facial, absence d’implantation pré-auriculaire des cheveux, présence de chondromes (tumeur bénigne du tissu cartilagineux) de la région prétragienne ou de la loge parotidienne.
- Nerveux : paralysie faciale (VII), parfois paralysie oculo-motrice (III, IV, VI).
- Une surdité de transmission dans près de 2/3 des cas.
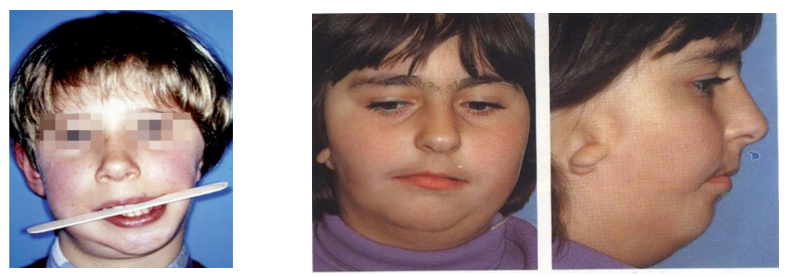
Quand l’atteinte est unilatérale, elle réalise le syndrome latéro-rétrognathie mandibulaire avec une déviation latérale du menton, une obliquité du plan d’occlusion vers le haut du côté atteint ainsi que des inclusions dentaires fréquentes. L’espace libre est considérablement réduit voire inexistant du côté atteint. Quand l’atteinte est bilatérale, on décrit une malocclusion de classe II avec parfois une béance, une étroitesse constante des voies aériennes par hypoplasie maxillaire, une fente palatine est présente dans 1/3 des cas, la parotide peut être absente ; l’intelligence est normale et le léger retard mental constaté par certains auteurs peut être secondaire à la surdité.
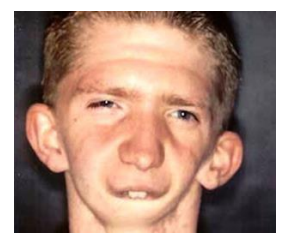
Autres noms de la maladie
- Syndrome du 1er arc branchial.
- Dysostose oto-mandibulaire.
- Micosomie crânio-faciale.
- Dysostose mandibulo-faciale.
- Syndrome de Treacher-Collins, de Franceschetti (atteinte bilatérale).
- Dysplasie zygo-auro-mandibulaire.
Fréquence
On retrouve 1 cas pour 3 500 naissances viables.
Étiopathogénie
Étiologie
C’est une maladie héréditaire, à transmission autosomique dominante avec expressivité variable et pénétrance de 100 %. Le gène muté responsable est le TCOF1.
Pathogénie
Cette neurocristopathie se traduit par des anomalies de la migration et de la division des cellules crestales provenant de la région du mésencéphale et du rhombencéphale et provoquant des délétions tissulaires au niveau de tous les dérivés du premier arc branchial.
Diagnostic
L’échographie 3D anténatale permettrait un diagnostic avant la naissance. Le diagnostic péri-natal est assez rare, sauf lorsque l’agénésie du pavillon de l’oreille est totale. Le diagnostic est en général posé dans les premiers mois de la vie, parfois plus tard, puisque la symptomatologie s’aggrave avec la croissance.
Traitement
Dans les formes unilatérales, deux schémas thérapeutiques peuvent être proposés, selon l’âge du diagnostic et surtout selon la date de la première consultation orthodontique.
Dans les cas vus précocement :
- Le traitement d’ODF est possible, dès l’âge de 6 à 8 ans par mise en place d’une plaque de surélévation interocclusale unilatérale, du côté opposé à la lésion. Cette surélévation va entraîner la constitution d’un espace libre entre les arcades du côté atteint et permettrait ainsi une éruption dentaire normale.
- Le traitement chirurgical comprend des interventions de chirurgie plastique pour reconstituer le pavillon de l’oreille.
- La correction chirurgicale de la latéro-gnathie par mise en place de greffon chondro-costal au niveau de la branche montante agénésique. Elle doit être différée jusqu’au stade de constitution de la denture adulte jeune, puisque c’est l’occlusion dentaire qui va être le guide de la correction chirurgicale.
Dans les cas vus plus tardivement :
- Lorsque la phase orthopédique n’a pu être réalisée, une normoclusion compensatrice et paradoxale s’est souvent installée. L’intervention chirurgicale majeure ne pourra donc pas avoir raisonnablement lieu avant une préparation orthodontique. Le schéma thérapeutique est donc le suivant :
- Traitement orthodontique préchirurgical, associant appareils multi-attaches avec tractions élastiques verticales et plaque de surélévation contro-latérale, destiné à corriger l’occlusion compensatrice.
- Les interventions de chirurgie plastique de reconstruction du pavillon ont lieu pendant cette période.
- Correction chirurgicale de la latéro-rétrognathie par greffe chondro-costale d’allongement mandibulaire, comme dans le schéma précédent.
- Traitement orthodontique post-opératoire et chirurgie plastique secondaire.
Achondroplasie
Définition / Description
L’achondroplasie est une maladie congénitale de l’os donnant un nanisme avec raccourcissement surtout de la racine des membres et un visage caractéristique. La transmission est de mode autosomique dominant, mais la majorité des cas sont le résultat de mutations spontanées du gène FGFR3 situé au niveau du chromosome 4. Ce gène est responsable de la synthèse du récepteur du facteur de croissance des fibroblastes (« fibroblast growth factor receptor »).
Diagnostic
Il est possible grâce à l’échographie du fœtus lors du troisième trimestre de la grossesse. De même à la naissance et après : le bébé a les membres courts et une tête disproportionnée ; un aplatissement de l’ensellure nasale, une saillie du maxillaire inférieur et un front large, les mains présentent un espace entre médius et annulaire : main en « trident », les membres inférieurs peuvent être incurvés. Les muscles paraissent volumineux.
Traitement
Consiste à traiter les complications neurochirurgicales et orthopédiques.
Conclusion
Tous les syndromes ne peuvent être cités. Il faut retenir que face à ces maladies, la prise en charge ne peut être que pluridisciplinaire. Le médecin dentiste veillera à maintenir la cavité buccale en état (dents et parodonte), l’orthodontiste traitera en tenant compte des spécificités de la maladie et de ses limites.
Bibliographie
- Philippe GUGNY : Guide illustré des malformations faciales les plus répandues. Rev Orthop Dento Faciale 24, 1990 : 439-463.
- B. Morand, E. Seigneuret, L. Selek, G. Bettega : Chirurgie des malformations craniofaciales : principes de base. EMC – Chirurgie orale et maxillo-faciale 1 Volume 11 n°3 août 2016.
- B Raphaël, J Lebeau, G. Bettega, JG Passagia, M Richard : Chirurgie des malformations craniofaciales. Encyclopédie médico-chirurgicale. 22-066-B-20.
Les grands syndromes en ODF / ODF
La santé bucco-dentaire est essentielle pour le bien-être général, nécessitant une formation rigoureuse et continue des dentistes. Les étudiants en médecine dentaire doivent maîtriser l’anatomie dentaire et les techniques de diagnostic pour exceller. Les praticiens doivent adopter les nouvelles technologies, comme la radiographie numérique, pour améliorer la précision des soins. La prévention, via l’éducation à l’hygiène buccale, reste la pierre angulaire de la pratique dentaire moderne. Les étudiants doivent se familiariser avec la gestion des urgences dentaires, comme les abcès ou les fractures dentaires. La collaboration interdisciplinaire avec d’autres professionnels de santé optimise la prise en charge des patients complexes. La santé bucco-dentaire est essentielle pour le bien-être général, nécessitant une formation rigoureuse et continue des dentistes.
Les grands syndromes en ODF / ODF
Articles similaires

Angine de Ludwig : Symptômes, Traitements et Prévention de cette Infection Grave
Angine de Ludwig : Symptômes, Traitements et Prévention de cette Infection Grave Imaginez une simple douleur dentaire qui se transforme...
Lire l'article
Prescription médicamenteuse chez l’enfant
Prescription médicamenteuse chez l’enfant INTRODUCTION La prescription médicamenteuse occupe une place importante dans la prise en charge de la pathologie...
Lire l'article
Obturation Thermoplastique : Tout Savoir sur cette Technique Moderne d’Endodontie
Obturation Thermoplastique : Tout Savoir sur cette Technique Moderne d’Endodontie Vous avez besoin d’un traitement de canal et votre dentiste...
Lire l'article
Leave a Reply