Les grands syndromes en ODF
Les grands syndromes en ODF
Introduction
les troubles de la morphogénèse peuvent avoir des répercussions sur le développement des structures oro- faciales donnant naissance des anomalies orthodontiques souvent associées à des malformations des autres organes.
L’objectif de ce cours est de passer en revue certaines de ces malformations ; les plus fréquentes que nous pouvons être amenées à rencontrer.
- Définitions :
-Syndrome : Un syndrome se définit comme un ensemble de plusieurs symptômes ou signes en rapport avec un état pathologique donné et permettant, par leur groupement, d’orienter le diagnostic.
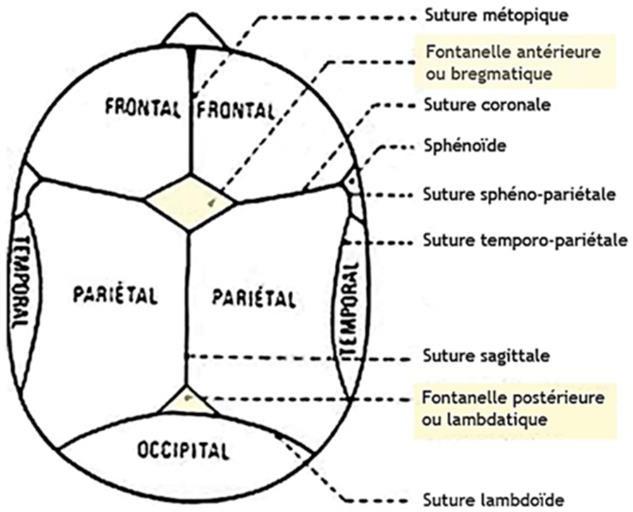
-Crâniosténose : La craniosténose est une anomalie primitive de croissance du squelette crânien associée à la fermeture prématurée d’une ou plusieurs sutures crâniennes. Il existe diverses formes de craniosténoses sans et avec atteinte du massif facial ou des extrémités (syndromes de Crouzon, Pfeiffer, Saethre-Chotzen, Apert…).
Les craniosténoses posent un problème :
- Morphologique : une dysmorphie crânienne, et faciale
- Fonctionnel : un conflit de croissance entre crâne et cerveau.
Cela peut avoir un retentissement mental, visuel et psychologique si le traitement n’est pas suffisamment précoce.
-Dysplasie : une dysplasie désigne toute anomalie du développement d’une cellule, d’un tissu ou d’un organe se traduisant par une déformation ou une malformation cellulaire,
tissulaire ou organique.
–La dysostose : désigne une déformation qui touche préférentiellement une ou plusieurs pièces osseuses.
- Les grands syndromes pathologiques à répercussions orthodontiques :
- Les crâniosténoses
- Syndrome de Crouzon ou la dysostose crânio-faciale de CROUZON :
- Définition – Description
- C’est la plus connue des craniosténoses, cette malformation crânio-faciale d’origine héréditaire a été décrite par CROUZON.
- L’étiologie est héréditaire en rapport avec une mutation du gène FGFR2 (ou
en anglais : fibroblast growth factor receptor 2 localisé sur le locus q26 du chromosome 10. Il existe deux allèles de cette mutation), autosomale dominante avec expressivité variable
– Pathogénie : c’est la conséquence de la fermeture
prématurée de la suture coronale, lambdoïde ainsi que des sutures sphéno-occipitale et sphéno-vomerienne et la
synostose entre le vomer et les maxillaires.
Figure 01 : Syndrome de CROUZON.
- Anomalies crânio-faciales :
- Bosse frontale.
- Exophtalmie avec strabisme divergent.
- Hypertélorisme.
- Augmentation de la pression intracrânienne.
- Impression digitiforme sur la voûte crânienne.
- Nez en bec de perroquet.
- Anomalies buccales :
- Hypoplasie du maxillaire supérieur avec DDM.
- Palais ogival.
- Prognathie mandibulaire.
- Oligodontie.
- Fente labio-palatine dans 10 à 15%.
- Le psychisme est primitivement normal et il n’y a pas d’arriération mentale.
- Fréquence : La prévalence dans la population générale en Europe est estimée à 1/50 000.
2.1.2 Syndrome d’APERT ou acro-céphalo-syndactylie :
C’est un syndrome plus rare que le syndrome de Crouzon.
- une malformation majeure, associant une facio-crânio-sténose et des syndactylies osseuses et membranaires des quatre extrémités.
- L’étiologie est héréditaire, elle se transmet selon un mode autosomique dominant mutation du gène FGFR2, mais la plupart des cas sont le plus souvent sporadiques.
- Pathogénie : c’est la fermeture prématurée de la suture coronale, lambdoide ainsi que des sutures sphéno-occipitale et sphéno-vomerienne et la synostose entre le vomer et les maxillaires.
- Le tableau clinique du syndrome d’Apert est très polyvalent, il comporte :
- Des malformations crânio-faciales :
- une diminution du diamètre antéropostérieur du crâne (brachycéphalie)
- une hypoplasie de l’étage moyen de la face avec un rétrécissement des loges orbitaires de degré variable responsable parfois d’un exorbitisme.
- Les orbites sont orientées en dehors et latéralement
- le nez est mince et pointu.
- Mains et pieds :
Une syndactylie avec fusion des os et de la peau des phalanges distales touchant les deux mains et pieds de façon égale (aspect en moufle des extrémités), permettant de le différencier des autres
syndromes (syndrome de Crouzon).
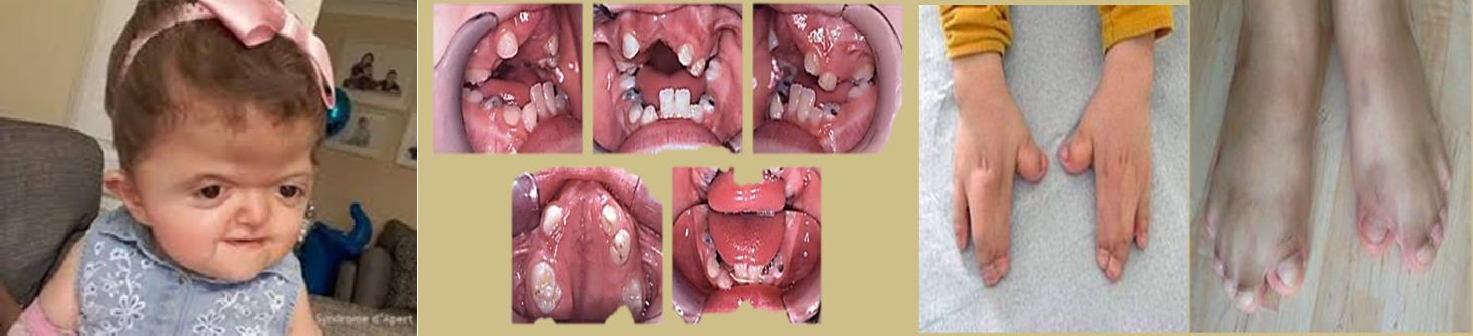
Figure 02 : syndrome d’APERT.
- Anomalies buccales :
- Hypoplasie du maxillaire avec pour conséquence une dysharmonie dento-maxillaire supérieure.
- Inversion de l’articulé dentaire (malocclusion de Cl III).
- Une division palatine très étroite en générale.
- Fréquence : elle est estimée approximativement 1 cas sur 65 000 naissances viables.
- Syndrome de Binder ou dysostose maxillo-nasale ou syndrome naso-maxillo-vertébral
- Définition – Description :
C’est un syndrome malformatif congénital très rare touchant essentiellement l’étage moyen de la face dont les caractéristiques principales sont :
- Nez plat et vertical.
- malposition des os propres du nez.
- Hypoplasie ou absence d’épine nasale antérieure.
- Atrophie de la muqueuse nasale.
- Absence de sinus frontaux.
- L’angle naso-frontal peut être plus obtus.
- Hypoplasie naso-maxillaire avec malocclusion de classe III
- Des malformations de la charnière cervico-occipitale et du rachis cervical, dans 50 % des cas.
- Fréquence : ce syndrome survient chez environ 1 nouveau-né sur 10 000.
- Étiologie : inconnue ; les experts ne savent pas exactement ce qui cause le syndrome de Binder. La plupart du temps, les bébés développent cette maladie sans raison connue.
Les chercheurs pensent que certains facteurs environnementaux pourraient augmenter le risque d’avoir un bébé atteint du syndrome de Binder. Ces facteurs comprennent :
- Consommation d’alcool pendant la grossesse.
- Exposition in utero à certains médicaments, dont la phénytoïne (Dilantin®, Phenytek®) ou la warfarine (Coumadin®, Jantoven®).
- Carence en vitamine K pendant la grossesse.
- Traumatisme subi par votre bébé lors de l’accouchement.
Figure 03 : syndrome de BINDER.
- Syndrome oto-mandibulaire ou syndrome du 1er arc branchial ou dysostose otomandibulaire ou microsomie crânio-faciale
- Définition – Description
C’est l’ensemble des malformations associant hypoplasie ou agénésie de l’oreille et hypoplasie mandibulaire. La malformation peut être uni- ou bilatérale et dans ce cas symétrique ou
asymétrique. Elle peut être isolée (comme le syndrome de Franceschetti) ou associée à d’autres malformations (comme syndrome de de Nager).
- Le syndrome de Franceschetti- Klein (ou syndrome de Treacher-Collins) est un syndrome génétique autosomique dominant caractérisé par l’existence d’une anomalie du développement
crânio-facial, avec atteintes bilatérales et asymétriques. Sa fréquence est de 1/50 000 naissances.
Ses signes majeurs sont les suivants :
- Une hypoplasie du pavillon des oreilles ;
- Une atrésie des conduits auditifs externes ;
- Une anomalie des osselets avec surdité de transmission ;
- Une hypoplasie des os malaires et zygomatiques avec obliquité antimongoloïde des fentes palpébrales et un colobome de la paupière inférieure avec absence de cils du tiers externe ;
- La mandibule est en rétrusion avec un menton osseux fuyant et une hypoplasie des ramus ;
- Fente palatine.
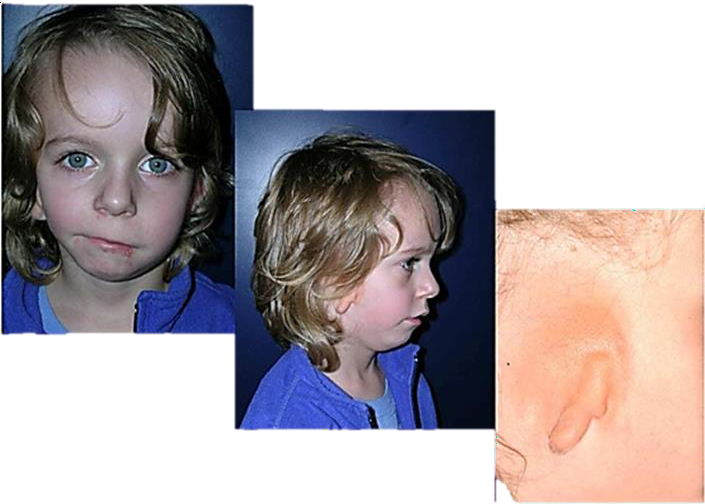
Figure 04 : syndrome de Franceschetti.
- Dysplasie cléidocrânienne (D. C. C.) ou syndrome de Pierre Marie et Sainton :
- Définition – Description
C’est une malformation génétique héréditaire rare, à transmission autosomique dominante, aux formes d’expressions variables.
Elle est caractérisée par :
- Anomalies crâniennes : un défaut de développement des os du crâne (larges sutures crâniennes), brachycéphalie, fermeture tardive des fontanelles et des bosses frontales saillantes.
- Anomalies faciales : l’hypoplasie du massif facial se traduit par palais profond et ogival (endognathie), une micro-rétrognathie maxillaire et un pseudo-prognathisme mandibulaire,
- Des anomalies dentaires : un retard ou une absence dans l’exfoliation des dents temporaires, l’inclusion des dents permanentes et la présence de dents surnuméraires.
- Anomalies des ceintures scapulaire et pelvienne : une aplasie ou une hypoplasie des clavicules, un retard de fermeture de la symphyse pubienne à l’âge adulte et une hypoplasie et une rotation antérieure des ailes et des articulations sacroiliaques.
- Anomalies thoraco-rachidiennes : un thorax en forme de cône, le rachis est incomplètement ossifié, des côtes et de vertèbres surnuméraires.
- Anomalies des membres supérieurs et inférieurs : une hypoplasie de l’humérus et du radius, coxa- vara, un genu valgum (ou genou en X).
– Fréquence : 1 cas sur 1 000 000.
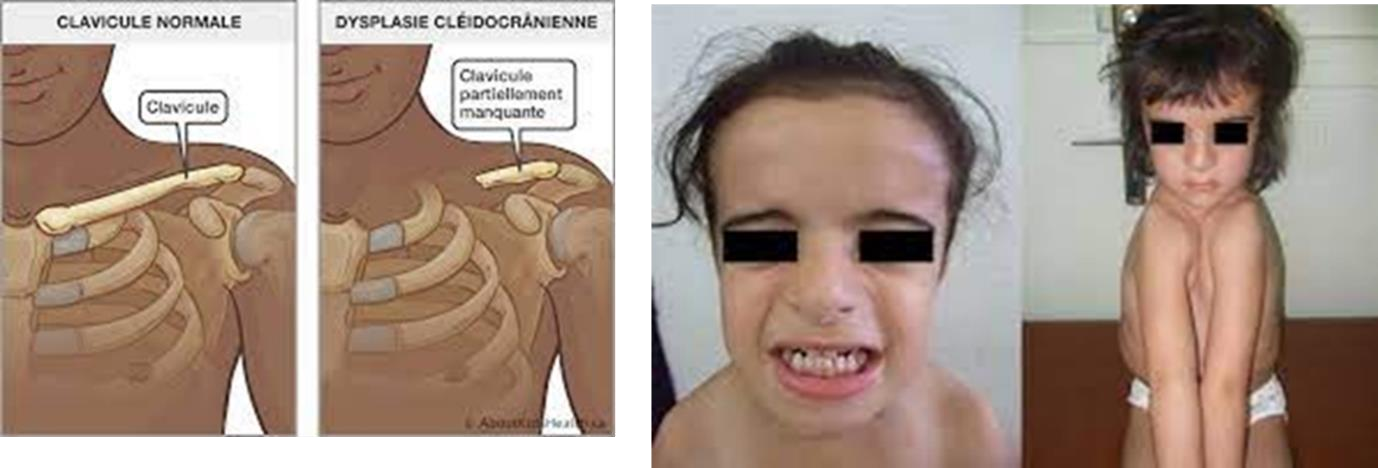
Figure 05 : Syndrome de Pierre Marie et Sainton.
- Syndrome de Pierre Robin (séquence de Pierre Robin) :
Syndrome crânio-facial, il peut être isolé, sporadique ou familiale. D’une incidence allant de 1/8 000 à 10 000.
Il est caractérisé par un tableau clinique de détresse respiratoire, et est définit par l’association de trois éléments :
- Rétrognathie mandibulaire,
- Glossoptose,
- Fente vélo-palatine.
- Etiologie : la séquence de Pierre Robin isolée est sporadique dans la majorité des cas une origine familiale.
- Anomalies buccales :
- Rétrognathie mandibulaire : profil « d’oiseau »
- Fentes palatines fréquentes.
- Glossoptose (chute de la langue en arrière, ce qui a pour conséquence de rétrécir le diamètre du pharynx avec risque d’obstruction des voies aériennes supérieures.
- Troubles de la déglutition et de la succion
- Anomalies associées :
- Troubles de la régulation respiratoire et cardiaque.
- Atrésie des oreilles.
- Fréquence :
Sa prévalence à la naissance est estimée à environ un nouveau-né sur 8 000 à 10 000.
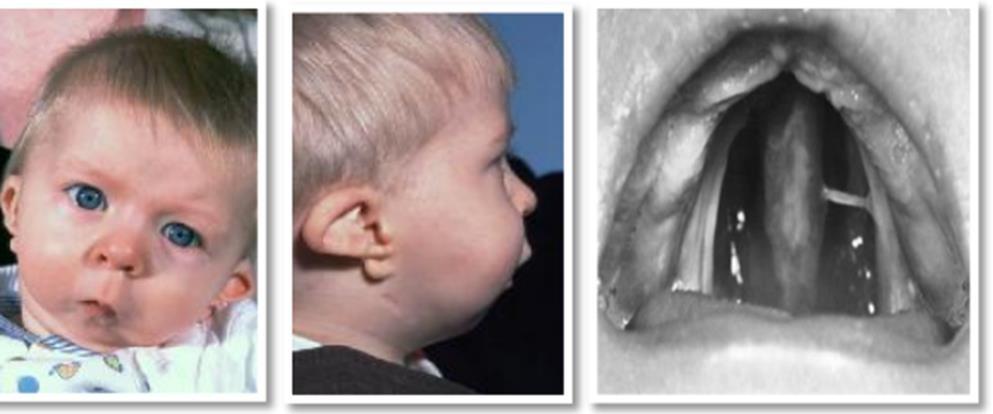
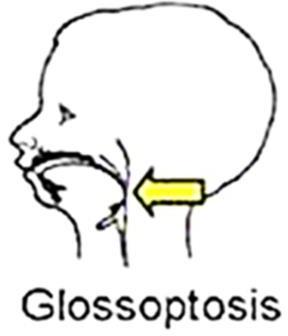
Figure 06 : syndrome de Robin.
- Syndrome de BRODIE « scissorsbite » :
C’est une anomalie transversale rare, caractérisée par une occlusion exagérée des secteurs latéraux, le contacte s’établissant entre les faces palatines des dents maxillaires et les faces vestibulaires des dents mandibulaires.
Cette affection présente de multiples conséquences ; dentaires, parodontales, fonctionnelles et articulaires auxquelles devra faire face l’orthodontiste.
Le syndrome de Brodie est rare (1,0% à 1,5%) dans la population générale. Ce syndrome demeure
extrêmement difficile à corriger et surtout la forme unilatérale chez l’adulte. Son approche diagnostique et thérapeutique est délicate.
Il s’agit d’une « occlusion vestibulaire complète de l’arcade supérieure » par une position haute de la langue (Brodie, 1952), qui peut être liée à :
-Un maxillaire hyper large (Brodie, Ramsay)
-Une mandibule rétrusive (Yogosawa).
- Une constriction de l’arcade mandibulaire (Bassigny, Guerrero, Alexander)
-Un trouble associé maxillaire et mandibulaire (Proffit)
De façon schématique, les moyens mis en œuvre vont chercher à contracter le maxillaire, et à élargir la
mandibule à un niveau alvéolaire et/ou basal dans le but de rétablir une occlusion postérieure fonctionnelle.
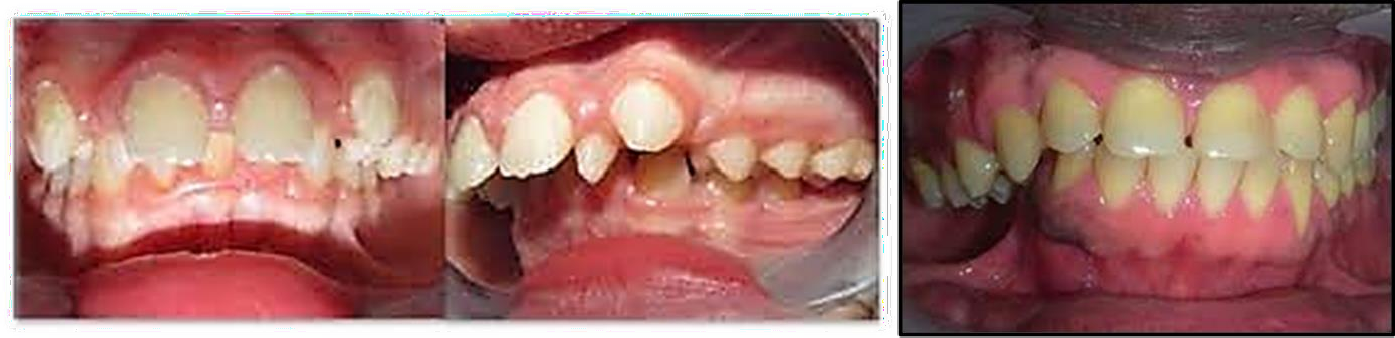
Figure 07 : Syndrome de BRODIE.
- Syndrome de Cauhépé-Fieux :
Il s’agit d’un syndrome purement orthodontique qui se caractérise par :
- Endoalvéolie supérieure,
- Une latérodéviation fonctionnelle,
- Un articulé croisé unilatéral (du côté de la déviation),
- Déglutition infantile et une interposition bilatérale de la langue.
Dans les formes graves : La symptomatologie est dominée par l’asymétrie du bas du visage avec :
- Malposition droite ou gauche de la mandibule,
- La ligne de symétrie est ainsi projetée d’un côté ou de l’autre,
- Les points dentaires supérieurs et inférieures ne concordent plus.
Nb : la mandibule est généralement normale pour sa forme.
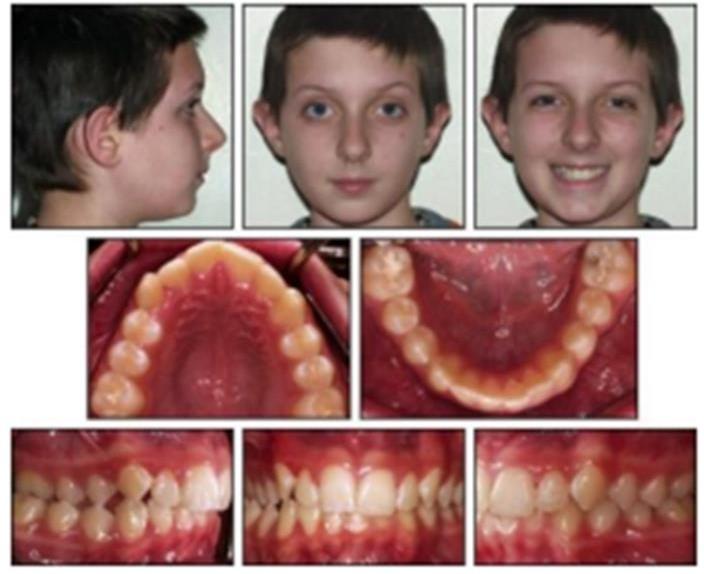
Figure 08 : syndrome de Cauhépé-Fieux.
Conclusion
L’orthodontiste a un rôle prépondérant dans le diagnostic de ces pathologies ce qui implique une bonne connaissance des signes, de l’étiologie et pathogénie de ces syndromes. La prise en charge nécessite une symbiose pluridisciplinaire.
Les grands syndromes en ODF
Voici une sélection de livres en français sur les prothèses dentaires:
Prothèse fixée, 2e Ed.: Approche clinique Relié – Illustré, 4 janvier 2024
Prothèse Amovible Partielle : Clinique et Laboratoire
Collège National des Enseignants en Prothèses Odontologiques (CNEPO), Michel Ruquet, Bruno Tavernier
Traitements Prothétiques et Implantaires de l’Édenté Total 2.0
Conception et Réalisation des Châssis en Prothèse Amovible Partielle
Prothèses supra-implantaires: Données et conceptions actuelles
Prothèse complète: Clinique et laboratoire Broché – Illustré, 12 octobre 2017
Les grands syndromes en ODF
Articles similaires

Les étapes de laboratoire de PPAM
Les étapes de laboratoire de PPAM Introduction La confection d’une prothèse partielle amovible métallique (PPAM) est un processus complexe qui...
Lire l'article
Accident d’Exposition au Sang (AES) en Cabinet Dentaire : Conduite à Tenir et Prévention
Les accidents d’exposition au sang (AES) représentent un risque professionnel majeur dans les cabinets dentaires. Chaque année en France, près...
Lire l'article
Dynamique de la Lésion Carieuse / Odontologie Conservatrice
Dynamique de la Lésion Carieuse / Odontologie Conservatrice Introduction La carie peut affecter l’émail, la dentine et le cément de...
Lire l'article
Leave a Reply