
Prise en charge des patients à besoins spéciaux : Hémopathies / PBD
Prise en charge des patients à besoins spéciaux : Hémopathies / PBD
Introduction
Les hémopathies sont des affections caractérisées par une atteinte des éléments figurés du sang. Seuls les examens de laboratoire permettent leur diagnostic.
Plan
- Introduction
- Rappels
- Étude clinique :
- Syndromes leucocytaires non prolifératifs
- Syndromes prolifératifs
- Syndromes anémiques
- Syndromes hémorragiques
Rappels
Rôle du sang
Le sang circule dans le système circulatoire, irrigue tous les tissus de l’organisme en apportant des substances nutritives, de l’énergie et de l’oxygène. Il recueille les déchets pour les transporter vers les reins, les poumons, etc., et participe à la défense contre les infections.
Composition du sang
Le sang se compose de deux parties :
- Plasma
- Éléments figurés : globules rouges (hématies), globules blancs (leucocytes), plaquettes (thrombocytes).
Système hématopoïétique
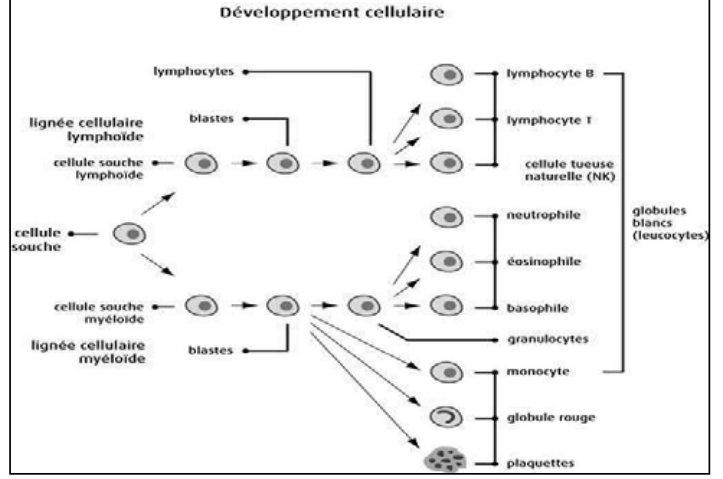
Il comprend la moelle osseuse, les ganglions lymphatiques et la rate.
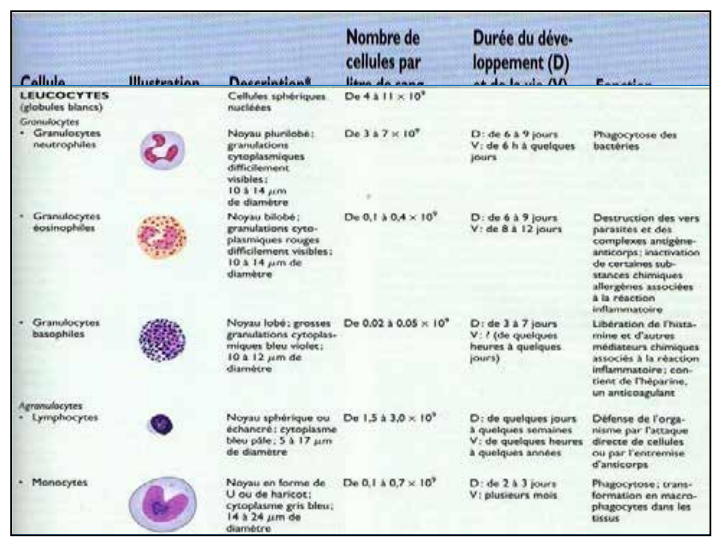
Caractéristiques des cellules sanguines
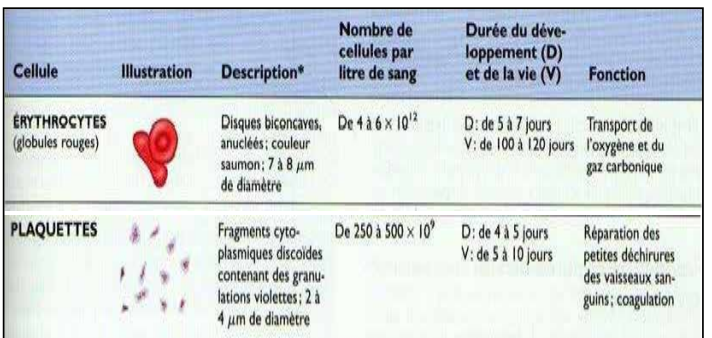
| Cellule | Description | Nombre de cellules par litre de sang | Durée de développement (D) et de vie (V) | Fonction |
|---|---|---|---|---|
| Érythrocytes (globules rouges) | Disques biconcaves non nucléés, couleur rouge, 7 à 8 µm de diamètre | 4 à 6 × 10¹² | – | Transport de l’oxygène |
| Plaquettes | Fragments cytoplasmiques accolés contenant des granulés violets, 2 à 4 µm de diamètre | 250 à 500 × 10⁹ | – | Hémostase |
Évaluation
- Numération des globules rouges par mm³ de sang
- Hématocrite (HT) : 40-48 %
- Taux d’hémoglobine (Hb) : 12-18 g/dL
Hémogramme
L’hémogramme comporte deux types d’analyses :
- Quantitative : numération formule sanguine (NFS).
- Morphologique : analyse des cellules.
Hémostase
L’hémostase intervient pour colmater les fuites dans le circuit vasculaire et maintenir le sang fluide à l’intérieur des vaisseaux. Elle fait intervenir la paroi vasculaire, les cellules sanguines et les protéines plasmatiques.
Hémostase primaire
- Vasoconstriction immédiate et adhésion des plaquettes.
- Agrégation réversible devenant irréversible (thrombus blanc).
Hémostase secondaire
- Prothrombine (prothrombinase) → thrombine.
- Fibrinogène → fibres insolubles (thrombus rouge).
- Voies :
- Intrinsèque : facteurs VIII, IX, XI et XII.
- Extrinsèque : facteurs II, V, VII et X.
Fibrinolyse
- Plasminogène → plasmine.
- Fibrine → produits de dégradation de la fibrine.
Exploration de l’hémostase
| Exploration | Tests | Normalités |
|---|---|---|
| Hémostase primaire | Temps de saignement (TS) | Duke : 2-4 min, Ivy : 4-8 min |
| Numération plaquettaire (NP) | 150 000-450 000/mm³ | |
| Coagulation | TCA/TCK, dosage des facteurs (voie intrinsèque et commune) | 30-40 secondes |
| TQ, TP, INR, dosage des facteurs (voie extrinsèque et commune) | TQ : 12-13 s, TP : 70-100 %, INR : 1 | |
| Fibrinolyse | Test de Von Kaulla (temps de lyse des euglobulines) | 3 heures |
Étude clinique et prise en charge thérapeutique
Syndromes leucocytaires non prolifératifs
Quantitatif
- Insuffisance : leucopénie.
- Excès : leucocytose.
Qualitatif
- Anomalie fonctionnelle.
Désordres lymphocytaires
Lymphocytose
Excès de lymphocytes, pouvant résulter de :
- Processus infectieux (ex. : mononucléose infectieuse).
- Désordres hématopoïétiques (ex. : leucémie lymphoblastique, lymphome, leucémie lymphoïde chronique).
Lymphopénies
- Déficit immunitaire primitif : lymphocytes B et T ou T et B.
- Déficit immunitaire secondaire :
- Infections (ex. : VIH affectant les lymphocytes T4).
- États inflammatoires, maladie de Hodgkin.
- Médicaments immunosuppresseurs, chimiothérapie, radiothérapie.
Désordres associés aux polynucléaires neutrophiles
Neutropénie
Diminution des polynucléaires neutrophiles :
- Discrète : 1 000 à 1 500 cellules/µL.
- Modérée : 500 à 1 000 cellules/µL.
- Sévère (agranulocytose) : < 500 cellules/µL.
Risque infectieux : important en dessous de 0,5 × 10⁹/L.
Causes :
- Centrales : défaut de production médullaire.
- Périphériques : prise médicamenteuse, maladies auto-immunes.
Diagnostic : numération des polynucléaires.
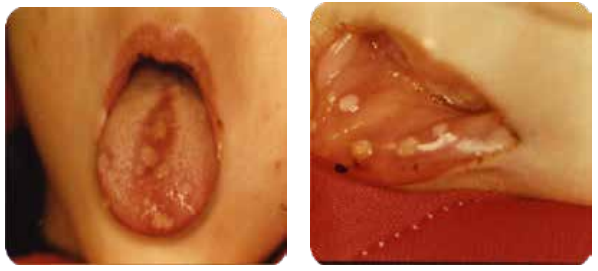
Manifestations buccales :
- Ulcérations semblables à des lésions aphtoïdes, douloureuses, entraînant dysphagie et sialorrhée.
- Infections : bactériennes (gingivite), virales (herpès), fongiques (candidoses).
Agranulocytose
Forme sévère de neutropénie (< 500 cellules/µL).
- Agranulocytose pure : état infectieux intense avec hyperthermie à 40 °C et frissons.
- Agranulocytose toxique/médicamenteuse : mécanisme immuno-allergique ou hypersensibilité (antimitotiques, sulfamides, arsenicaux, barbituriques).
- Localisation : ulcérations postérieures au voile du palais et à l’oropharynx, entraînant sialorrhée et dysphagie.
Agranulocytoses constitutionnelles (rares) :
- Agranulocytose génétique infantile.
- Neutropénie sévère congénitale.
- Gingivite dès l’éruption des dents temporaires, ulcérations rebelles, parodontites aiguës.
- Évolution possible vers une leucémie aiguë myéloblastique.
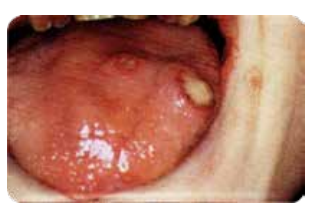
Atteinte qualitative
Dysfonctions des polynucléaires neutrophiles (action phagocytaire).
Origine héréditaire :
- Syndrome de Chédiak-Higashi : atteinte quantitative et qualitative des granulocytes.
- Ulcérations buccales, exfoliation prématurée des dents temporaires, gingivites, parodontites sévères liées au déficit immunitaire.
Origine acquise :
- Médicaments (aspirine, ibuprofène, corticoïdes).
- Intoxication éthylique.
Polynucléose à éosinophiles
Manifestation secondaire à :
- Néoplasies, lymphome, maladie de Hodgkin.
- Allergies, maladies du collagène vasculaire, parasitoses.
- Causes : allergiques (eczéma, urticaire), parasitaires.
Polynucléose à basophiles
Rare, sauf dans les syndromes myéloprolifératifs chroniques.
Prise en charge
- Avis du médecin traitant.
- Réduire le stress lors des soins, privilégier des soins de courte durée avec sédation.
- Pas de contre-indication aux vasoconstricteurs ; injections lentes après aspiration.
- Éliminer toutes sources d’infection.
- Hygiène bucco-dentaire rigoureuse.
- Antibioprophylaxie avant tout acte chirurgical en cas de neutropénie ou lymphopénie.
- Mesures d’hygiène et d’asepsie strictes.
- Précautions lors des prescriptions : éviter les médicaments dépresseurs de la moelle osseuse.
- Prendre en compte les pathologies associées.
Syndromes prolifératifs
Maladie de Vaquez
Polyglobulie vraie/primitive.
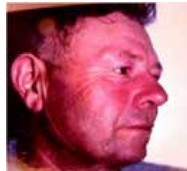
Hémogramme :
- Hématies : 6 à 8 millions/mm³.
- Hématocrite : > 55 % (femme), > 60 % (homme).
- Hémoglobine : > 17 g/dL (homme), > 16 g/dL (femme).
- Hyperleucocytose, hyperplaquettose (> 1 000 000/mm³).
Symptômes :
- Céphalées, vertiges, fatigue.
- Érythrose particulière du visage.
Manifestations buccales :
- Muqueuse buccale rouge, violacée, œdématiée.
- Pétéchies, ulcérations, gingivorragies spontanées.
- Voile du palais rouge pourpre.
- Complications liées à des thromboses ou troubles de l’hémostase.
Leucémies
Cancer affectant les globules blancs, dû à des anomalies chromosomiques, irradiations ou expositions chimiques.
Classification :
- Aiguë/chronique.
- Lymphoïde/myéloïde.
Diagnostic :
- Myélogramme : envahissement médullaire par des leucoblastes.
- Analyse cytologique : type cellulaire (lymphoblastique/myéloblastique).
Leucémies aiguës
Prolifération de blastes immatures envahissant la moelle et parfois le sang.
Leucémies chroniques
- Leucémie myéloïde chronique (LMC) :
- Peu de signes buccaux sauf en cas d’aplasie médullaire thérapeutique.
- Leucémie lymphoïde chronique (LLC) :
- Hyperlymphocytose, polyadénopathie, adénopathies cervicales, splénomégalie.
- Risque de tumeur maligne buccale ou ORL.
- Diagnostic :
- NFS : hyperlymphocytose (> 4 000/mm³) ± adénopathie.
- Frottis sanguin : petits lymphocytes matures normaux.
Manifestations buccales :
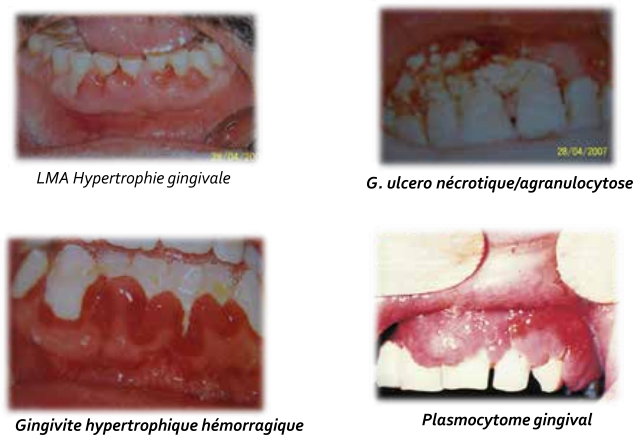
- Liées à :
- Insuffisance médullaire.
- Syndrome tumoral.
- Chimiothérapie.
- Insuffisance médullaire :
- Anémie : asthénie, tachycardie, pâleur muqueuse.
- Neutropénie : ulcérations nécrotiques, syndromes fébriles, foyers infectieux dentaires.
- Thrombopénie (< 30 000/mm³) : gingivorragies, pétéchies, épistaxis.
- Syndrome tumoral :
- Adénopathies cervicales, infiltration des glandes salivaires.
- Gingivite hypertrophique hémorragique, ulcérations, douleurs.
- Algies dentaires par infiltration pulpaire.
- Chimiothérapie :
- Toxicités : hémorragies, xérostomie, adénopathies, pharyngites, gingivorragies, infections récurrentes, érosion alvéolaire.
- Coloration violette des muqueuses (misulban).
- Corticothérapie : œdèmes (Cushing).
- Greffe de moelle : réaction du greffon contre l’hôte (lichen plan).
Prise en charge
- Consultation médicale si antécédents ou symptômes de leucémie.
- Éviction des foyers infectieux chroniques.
- Hémostase locale si plaquettes < 100 000/mm³.
- Chimiothérapie :
- Lutte contre les mucites : hygiène bucco-dentaire, bains de bouche (bétadine), pansements digestifs, laser thérapie.
- Infections bactériennes : antibiotiques.
- Prévention des ulcérations : acide folique IV ou topique (3 fois/jour).
- Infections fongiques : nystatine orale (10 mL, 100 000 UI, 4 fois/jour).
- Période d’aplasie profonde :
- Prévention des infections exogènes/endogènes : antibiotiques, bains de bouche (mycostatine, lidocaïne, bétadine, bicarbonate).
- Contre-indication aux gestes chirurgicaux.
- Urgences : intervention en milieu hospitalier avec hématologue.
- Phase de début :
- Tuméfaction, douleurs, fièvre : consultation hématologique.
- Urgences : antibiotiques, hémostase, hospitalisation si plaquettes < 80 000/mm³.
- Phase de rémission :
- Remise en état buccale, élimination des sources d’infection.
- Prophylaxie antifongique (nystatine, 10 mL, 10 000 UI, 4 fois/jour).
Lymphomes et myélomes multiples
Myélome multiple (maladie de Kahler)
Prolifération maligne de plasmocytes sécrétant des immunoglobulines monoclonales.
Signes :
- Douleurs osseuses, tuméfactions, fractures.
- Anémie, infections, hémorragies, insuffisance rénale.
Manifestations buccales :
- Lésions péri-apicales (molaire, prémolaire mandibulaire, ramus, angle).
- Lésions nodulaires des tissus mous.
- Fractures pathologiques, gingivorragies, pétéchies.
Lymphomes
Maladie de Hodgkin
- Cellules de Sternberg dans les organes atteints.
- Prédilection pour les ganglions cervicaux inférieurs.
- Localisations initiales : amygdaliennes, linguales, salivaires.
- Signes : anémie, hémorragies, infections, déficit immunitaire.
Lymphomes non hodgkiniens
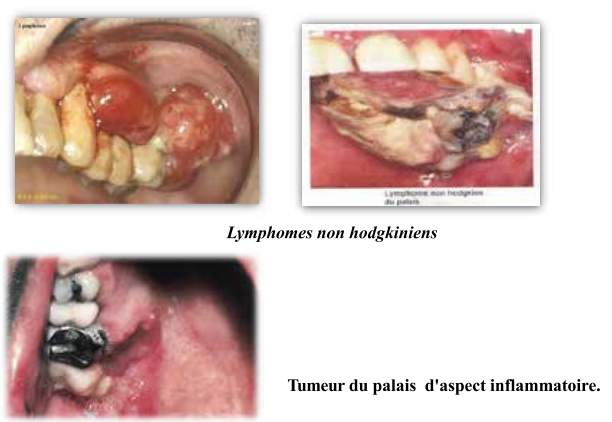
- Proliférations plasmocytaires monoclonales malignes (organes lymphoïdes secondaires).
- Deux fois plus fréquents que Hodgkin.
- Signes maxillo-faciaux : adénopathie cervico-faciale ferme, élastique, indolore, unique ou multiple.
- Tumeur du palais d’aspect inflammatoire.
Prise en charge
- Dépistage : avis médical, travailler hors épisodes aigus.
- Précautions :
- Insuffisance médullaire (anémie, leucopénie, thrombopénie).
- Couverture antibiotique si globules blancs < 2 500/mm³.
- Maîtrise de l’hémostase locale.
- Précautions vis-à-vis de l’irradiation cervico-faciale.
Syndromes anémiques
Définition
Diminution de l’hémoglobine altérant le transport de l’oxygène, confirmée par :
- Hémogramme :
- Homme : < 13 g/dL.
- Femme/enfant : < 12 g/dL.
- Enfant < 1 an : < 11 g/dL.
- Nouveau-né : < 14 g/dL.
- Femme enceinte (3e trimestre) : < 10,5 g/dL.
- Baisse des globules rouges et de l’hématocrite.
Paramètres
- Volume globulaire moyen (VGM) : macrocytaire/microcytaire.
- Concentration corpusculaire moyenne en hémoglobine (CCMH) : normo/hypochrome.
- Teneur globulaire moyenne en hémoglobine (TGMH).
- Taux de réticulocytes.
- Taux de fer sérique.
Mécanismes
- Central : insuffisance de production médullaire.
- Périphérique : raccourcissement de la durée de vie des hématies (hémorragies, hyperhémolyse).
Insuffisance médullaire
- Quantitative : défaut de cellule souche (aplasie médullaire).
- Qualitative : moelle normale, cellules anormales.
Sémiologie
- Symptôme stomatologique : pâleur des muqueuses.
- Signes cutanéo-muqueux : pâleur cutanée, décoloration des muqueuses/conjonctives.
- Symptômes généraux : tachycardie, céphalées, vertiges, pertes de connaissance, fatigabilité, irritabilité.
Classification
Anémies normocytaires normochromes
- Anémies post-hémorragiques :
- Traitement : tarir l’hémorragie, transfusion, oxygénothérapie.
- Anémies hémolytiques congénitales :
- Ictère hémolytique (Minkowski-Chauffard) :
- Couverture antibiotique pour chaque intervention.
- Drépanocytose :
- Hémoglobinopathie déformant les globules rouges (fragiles, rigides, obstruant les vaisseaux).
- Hémogramme : anémie (Hb 7-9 g/dL), normochrome, normocytaire, régénérative.
- Signes : pâleur, ictère, algies osseuses, infections graves.
- Manifestations buccales : ostéoporose maxillaire, pâleur/jaunisse muqueuse, hypoplasie émail/dentine, douleurs mandibulaires, paresthésie labiale.
- Prise en charge :
- Avis médical, hospitalisation en cas d’infection.
- Antibioprophylaxie à large spectre (IV si possible).
- Ictère hémolytique (Minkowski-Chauffard) :
- Anémies hémolytiques acquises :
- Étiologies : allergie (pénicilline), affections hématologiques/immunologiques/infectieuses/cancéreuses.
- Traitement : antibioprophylaxie.
Anémies microcytaires hypochromes
- Thalassémie :
- Désordre génétique (diminution de synthèse de globine).
- Diagnostic : électrophorèse de l’hémoglobine.
- Thalassémie mineure (alpha) :
- Biochimie : hémolyse (bilirubine libre ↑), fer sérique ↑, ferritinémie ↑.
- Traitement : transfusion.
- Thalassémie majeure (bêta, anémie de Cooley) :
- Hémogramme : anémie (4-7 g/dL), microcytaire (60-70 fL), hypochrome, réticulocytes normaux.
- Manifestations buccales : hypertrophie/prognathie maxillaire, inflammation gingivale, anomalies dentaires, trabéculation lâche (radiographie).
- Prise en charge :
- Avis médical, évaluation Hb/Ht.
- Conséquences : arythmie, insuffisance myocardique, fibrose/cirrhose hépatique.
- Hb 8-10 g/100 mL : soins palliatifs.
- Antibioprophylaxie en concertation.
- Anémies sidéropéniques/ferriprives :
- Causes : insuffisance alimentaire, malabsorption (Crohn), hémorragies chroniques.
- Diagnostic : globules rouges rarement < 3 millions/mm³, fer sérique ↓, VGM 55-75 fL, CCMH 25-30 %.
- Traitement : fer ferreux, suppression de la cause.
- Précautions : intervenir si Hb > 10 g/100 mL, risque de retard de cicatrisation.
Anémies macrocytaires/mégaloblastiques
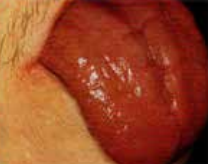
- Anémie de Biermer :
- Carence en vitamine B12, sujets âgés (surtout femmes).
- Déficit de facteur intrinsèque (Castle).
- Symptômes : troubles digestifs (nausées, diarrhées).
- Diagnostic : VGM ↑, Hb ↓, TGMH ↑, myélogramme (érythroblastes, myélocytes, mégacaryocytes), réticulocytes normaux/abaissés.
- Traitement : vitamine B12.
- Manifestations buccales :
- Glossite de Hunter (atrophie linguale douloureuse).
- Érosions aphtoïdes/fissuraires douloureuses.
Syndromes hémorragiques
Définition
Défaillance des mécanismes d’hémostase, se manifestant par :
- Hémorragies libres.
- Hémorragies sous-muqueuses : purpura, pétéchies, ecchymoses.
Examens biologiques
- Numération plaquettaire, TS, TCA, TQ (TP).
Maladies de l’hémostase primaire
- Atteinte vasculaire : purpura vasculaire, anomalies capillaires (pétéchies, ecchymoses).
- Atteinte plaquettaire :
- Quantitative : thrombopénie (< 100 000/mm³, centrale/périphérique).
- Qualitative : thrombopathie (congénitale/acquise).
Manifestations :
- Pétéchies, ecchymoses, hématomes.
- Allongement du temps de saignement.
Hémorragies par altération vasculaire
- Purpura : lésion hémorragique rouge pourpre, ne s’effaçant pas à la vitropression.
- Purpura rhumatoïde : capillarite inflammatoire, arthralgies, douleurs abdominales (allergie).
- Purpura vasculaire infectieux :
- Pétéchial sans hémorragie muqueuse (aigu).
- Ecchymotique, extensif, hémorragique (suraigu, contexte septique).
- Diagnostic : clinique, hémogramme/bilan d’hémostase normaux.
- Conduite :
- Consultation hématologique, examens biologiques.
- Éradication des foyers infectieux.
- Évaluation de la résistance capillaire.
- Soins urgents en accord avec le médecin, arrêt des médicaments causaux.
Syndromes hémorragiques par atteinte plaquettaire
- Thrombopénies :
- Origine : centrale (aplasie médullaire), périphérique (destruction).
- Biologie : plaquettes ↓, TS ↑, irrétractibilité du caillot, TC/TQ/fibrinémie normaux.
- Signes :
- Cutanés : purpura ecchymotique/pétéchial, hémorragies rares (digestives, cérébrales).
- Buccaux : pétéchies, vésicules hémorragiques, gencives gonflées/friables, saignements spontanés.
- Conduite :
- Modérée (50 000-100 000/mm³) : transfusion plaquettaire, hémostase locale.
- Sévère (< 50 000/mm³) : risque hémorragique élevé.
- Post-chimiothérapie : reporter l’acte.
- Tumorale réfractaire : transfusion, milieu hospitalier.
- Thrombopathies :
- Anomalies fonctionnelles (myéloprolifératif, médicamenteux).
- Conduite :
- Intervention hospitalière.
- Transfusion plaquettaire, hémostase locale.
- Corticoïdes : antibioprophylaxie chirurgicale.
- Immunosuppresseurs : attendre la fin du traitement.
- Proscrire aspirine, AINS.
Maladie de von Willebrand
Anomalie quantitative/qualitative du facteur von Willebrand (transport facteur VIII, adhésion plaquettaire).
Fréquence : 10-15/10 000 habitants, hommes et femmes.
Biologie :
- TS allongé, adhésivité/agrégation plaquettaire ↓, facteur VIII ↓ (TCA allongé).
- Dosage immunologique (F.vW:Ag).
Types :
- Type I (75 %) : déficit quantitatif partiel.
- Type II (20 %) : déficit qualitatif.
- Type III (rare) : déficit quantitatif complet.
Manifestations buccales :
- Purpura ecchymotique, gingivorragies spontanées, hémorragies amygdaliennes.
Conduite :
- Prévention : vérifier vWF RCo et F VIII.
- Chirurgie urgente : perfusion facteur VIII, puis von Willebrand après normalisation F VIII.
- Chirurgie programmée : perfusion vWF (sans F VIII) 12-24 h avant.
- Hémostase locale, acide tranexamique (per os, rinçage passif).
Troubles de la coagulation (hémostase secondaire)
- Sévérité :
- Sévère : facteur < 1 %.
- Modérée : 1-5 %.
- Mineure : 6-25 %.
- Tests : NP/TQ/TS normaux, TCA/dosage facteur anormal.
Hémophilie
Maladie héréditaire récessive (chromosome X).
Types :
- Hémophilie A : déficit facteur VIII (80-85 %).
- Hémophilie B : déficit facteur IX (15-20 %).
Signes :
- Hémorragies externes (épistaxis, buccales, digestives).
- Internes : ecchymoses, hématomes, hémarthroses.
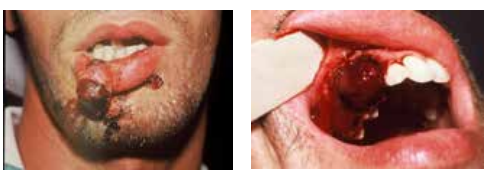
Traitement préventif :
- Hygiène bucco-dentaire, soins précoces.
- Proscrire injections IM, anesthésies tronculaires, aspirine, anti-hémostatiques.
- Injection du facteur déficient (plasmatique/recombinant).
Déficit acquis (facteurs vitamine K-dépendants)
Causes :
- Immaturité hépatique (nouveau-né).
- Anomalies synthèse/absorption vitamine K (antibiothérapie, maldigestion).
- Affections hépatobiliaires, éthylisme, anti-vitamines K.
Anti-vitamines K (AVK) :
- Inhibent facteurs II, VII, IX, X.
- Indications : maladies thromboemboliques, artériopathies, prothèses cardiaques, infarctus, AVC emboliques.
Signes cliniques :
- Ecchymoses, hémarthroses, pétéchies, vésicules hémorragiques, hyperplasies gingivales, gingivorragies.
Conduite :
- Interrogatoire : antécédents hémorragiques, médicamenteux, familiaux, pathologies associées.
- Tests : hémogramme, hémostase, sérologies post-transfusion.
- Anesthésie :
- Loco-régionale contre-indiquée (risque hématome latéro-pharyngé).
- Locale préférable (déficience légère/modérée).
- Patients sous anticoagulants :
- Vérifier TP-INR.
- Ne pas arrêter les anticoagulants.
- Sutures, colle chirurgicale, compresses hémostatiques.
- Traitement substitutif (concentrés de facteurs), hémostase locale, acide tranexamique (500 mg, 2 min, 4 fois/jour).
- Jeûne 2 h, boissons froides, alimentation légère.
Conclusion
- Rôle de dépistage : aucun signe pathognomonique.
- Examens biologiques : primordiaux pour le diagnostic et la prise en charge.
Prise en charge des patients à besoins spéciaux : Hémopathies / PBD
La santé bucco-dentaire est essentielle pour le bien-être général, nécessitant une formation rigoureuse et continue des dentistes. Les étudiants en médecine dentaire doivent maîtriser l’anatomie dentaire et les techniques de diagnostic pour exceller. Les praticiens doivent adopter les nouvelles technologies, comme la radiographie numérique, pour améliorer la précision des soins. La prévention, via l’éducation à l’hygiène buccale, reste la pierre angulaire de la pratique dentaire moderne. Les étudiants doivent se familiariser avec la gestion des urgences dentaires, comme les abcès ou les fractures dentaires. La collaboration interdisciplinaire avec d’autres professionnels de santé optimise la prise en charge des patients complexes. La santé bucco-dentaire est essentielle pour le bien-être général, nécessitant une formation rigoureuse et continue des dentistes.
Prise en charge des patients à besoins spéciaux : Hémopathies / PBD
Articles similaires

Prévention des maladies parodontales
Prévention des maladies parodontales Les maladies parodontales sont un problème de santé publique. Les médecins-dentistes et les pouvoirs publics ont...
Lire l'article
Bagues Transparentes ou Métal : Laquelle Choisir pour Redresser Vos Dents ?
Vous envisagez un traitement orthodontique, mais vous hésitez entre les bagues transparentes et les bagues métalliques ? Vous n’êtes pas...
Lire l'article
LES ECHECS EN IMPLANTOLOGIE
LES ECHECS EN IMPLANTOLOGIE « L’échec est le fondement du succès » (Lao Tseu) Introduction Quel que soit la fiabilité...
Lire l'article
Leave a Reply